
the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Jul 2019
| 26 Jul 2019
Potential of TLR-gene diversity in Czech indigenous cattle for resistance breeding as revealed by hybrid sequencing
Marek Bjelka
Kalifa Samake
Terezie Valčíková
A production herd of Czech Simmental cattle (Czech Red Pied, CRP), the conserved subpopulation of this breed, and the ancient local breed Czech Red cattle (CR) were screened for diversity in the antibacterial toll-like receptors (TLRs), which are members of the innate immune system. Polymerase chain reaction (PCR) amplicons of TLR1, TLR2, TLR4, TLR5, and TLR6 from pooled DNA samples were sequenced with PacBio technology, with 3–5× coverage per gene per animal. To increase the reliability of variant detection, the gDNA pools were sequenced in parallel with the Illumina X-ten platform at low coverage (60× per gene). The diversity in conserved CRP and CR was similar to the diversity in conserved and modern CRP, representing 76.4 % and 70.9 % of its variants, respectively. Sixty-eight (54.4 %) polymorphisms in the five TLR genes were shared by the two breeds, whereas 38 (30.4 %) were specific to the production herd of CRP; 4 (3.2 %) were specific to the broad CRP population; 7 (5.6 %) were present in both conserved populations; 5 (4.0 %) were present solely for the conserved CRP; and 3 (2.4 %) were restricted to CR. Consequently, gene pool erosion related to intensive breeding did not occur in Czech Simmental cattle. Similarly, no considerable consequences were found from known bottlenecks in the history of Czech Red cattle. On the other hand, the distinctness of the conserved populations and their potential for resistance breeding were only moderate. This relationship might be transferable to other non-abundant historical cattle breeds that are conserved as genetic resources. The estimates of polymorphism impact using Variant Effect Predictor and SIFT software tools allowed for the identification of candidate single-nucleotide polymorphisms (SNPs) for association studies related to infection resistance and targeted breeding. Knowledge of TLR-gene diversity present in Czech Simmental populations may aid in the potential transfer of variant characteristics from other breeds.
- Article
(1350 KB) - Full-text XML
- BibTeX
- EndNote





Resistance breeding is a prospective tool for the prevention of increases in infectious diseases in production populations of dairy cattle. This trend is considered to be a consequence of the increasing physiological load, unintended co-selection, and inbreeding associated with preferential breeding for milk production (Boichard et al., 2015). It also results from the emergence of new diseases (Purse et al., 2005) that are often resistant to standard antibiotic treatment.
In spite of the general adoption of genomic selection, an approach that targets causal genes might be more efficient in specific cases because the full available genetic variation is used. Disease-related breeding values are often underrepresented in combined breeding indices and the efficiency of current genomic selections might be disturbed by specific configurations in haplotype blocks (Abdel-Shafy et al., 2014).
The obvious targets for resequencing are components of the innate immune system (Boichard et al., 2015). In contrast to those of adaptive immunity they are fully determined in the germ line and are not formed ontogenetically (Kawai and Akira, 2010). A central role is played by the pattern-recognizing receptors (PRRs) that are activated by ligands originating in pathogenesis. The primary class of PRRs are the toll-like receptors, which are encoded by 10 gene paralogs denoted TLR1–TLR10 in cattle (toll-like receptors; Jungi et al., 2011). The so-called antibacterial series, comprising proteins TLR1, -2, -4, -5, and -6, recognize molecules from the cell walls of gram-negative and gram-positive bacteria. Notably, TLR5 recognizes the protein flagellin from bacterial flagella.
Natural variation in the TLR genes of cattle has been characterized across the panel of world breeds (Jann et al., 2008; Seabury et al., 2010; Fisher et al., 2011). Many of the variants, particularly the non-synonymous ones that code for an amino acid (aa) change in the coded product, allow for prediction of the consequences to the function of the corresponding receptor. For example, the well-known mutation 9787C > T (c.2021) in the TLR4 product is associated with a change in polarity in the crucial transmembrane part of the molecule, thereby affecting signal transfer (White et al., 2003). In accordance with expectations, this predicted functional change led to a shift in somatic cell count in cattle populations (Sharma et al., 2006; Beecher et al., 2010).
The importance of conserved historical populations of farm animals as a source of genetic diversity for resistance breeding is often acknowledged (FAO, 2007). Although the role of conserved populations in the protection of historical breeds is plain, gene pool erosion because of limited population sizes may significantly reduce their practical application in breeding; however, direct comparisons of the genetic richness in immunity genes with the modern production populations remain limited (Bilgen et al., 2016). For example, in historical Czech Red cattle previous studies reported high diversity in the major histocompatibility complex receptors (Hořín et al., 1997) and provided insight into the diversity of TLR4 (Novák et al., 2017).
Screening for variations in antibacterial members of the TLR gene family was conducted for Czech Simmental (Czech Red Pied, CRP) cattle to explore their potential use in breeding with the goal of disease resistance. To estimate the value of historical breeds as genetic resources for CRP, two related conserved populations were studied.
The Czech Red Pied breed was developed in the territory of Bohemia and Moravia during the 19th century from the imported original Simmental cattle. Breed book registration of bulls of the Fleckvieh and Montbélliard breeds began in 2000; therefore, the current trend leads to the convergence of gene pools with the major Simmental breeds. For conservation purposes, a nucleus herd was formed in 2010 from 70 animals corresponding to the gene pool prevailing at the end of the 1990s (Mátlová, 2013).
The CRP breed is considered to be dual purpose. The female population consists of 212 000 cows, representing 63.3 % of the female Holstein population and 32.7 % of the total cow population in the country. The difference in milk production compared to the Holstein breed persists; the CRP breed produces 7137 kg yr−1 compared to 9426 kg yr−1 in Holsteins (Czech-Moravian Breeder's Corporation, summarized data of 2017). On the other hand, the milk fat and protein contents are reliably more favourable in CRP. Regarding the proportions of the population, 40–70 bulls are evaluated for breeding value every year.
The third studied population was the conserved population of Czech Red cattle. The breed is sometimes tracked back to the ancient Celtic cattle of Roman times, analogous to other red highland cattle breeds of central and western Europe such as Harz Mountain cattle and Salers; however, molecular data for independent conclusions have yet to be collected (Ludwig et al., 2016). Czech Red cattle prevailed until the 18th century in the territory of Bohemia and Moravia and presumably contributed to the formation of the Czech Simmental. The actual herd was restored in 1978 from only 14 cows and one bull with 50 % identity (http://www.fao.org/dad-is/en, last access: 15 July 2019). The current number of animals in the conserved population is approximately 250.
To avoid the necessity of validation of the revealed polymorphisms using individual genotyping reactions, the principle of hybrid resequencing was applied. Dataset noise was mostly suppressed by combining two technologies for population resequencing. In addition to polymerase chain reaction (PCR) amplicons that were prepared from pooled DNA samples and sequenced with PacBio technology with 3–5× coverage per gene per animal, the gDNA pools were sequenced with the Illumina X-ten platform at low coverage. In the next step, the functional consequences for the identified non-synonymous variants were predicted. Knowledge of gene variants occurring in the involved breeds allows for a careful transfer of information about their contribution to disease resistance from previous studies in other breeds.
2.1 Input DNA samples
The primary collection of DNA samples was from 150 bulls in the Czech Red Pied production population. DNA was prepared from cryo-preserved insemination doses using the MagSep tissue method (Eppendorf, Hamburg, Germany). Insemination doses of 100 µL were incubated with lysis buffer and 25 µL of proteinase K from the MagSep kit in the presence of 80 mM dithiothreitol at 55 ∘C for 12 h with occasional vortexing prior to isolation.
The two conserved populations were characterized using DNA isolated from the aliquots of compulsory blood samples provided to the gene bank of the genetic resources programme. Thirty-five animals of the conserved CRP and 80 animals of CR (Czech Red) were included. One hundred microlitres of thawed blood was processed in silica membrane columns using the BloodPrep commercial procedure (Life Technologies, Carlsbad, CA, USA).
The concentration of the isolated DNA was determined fluorometrically on a microplate with SybrGold stain (Biotium, Fremont, CA, USA) and camera G:BOX Chemi XR5 (Syngene, Cambridge, UK) equipped with an EtBr filter under blue light excitation. Normalized genomic DNA (20 ng µL−1) was used as a template for PCR amplification of the exonic and flanking regions of all five antibacterial TLR genes. The amplification primers largely corresponded to those in previously published studies (White et al., 2003; Seabury et al., 2007; Seabury and Womack, 2008) and are listed in Table 1; the optimized PCR conditions are also listed. The PCR product yield was estimated according to the agarose electrophoresis control with GelRed (Biotium) fluorescent staining and a mixed sample was prepared containing all PCR fragments in equimolar concentration. The amplicon pool was purified on NucleoSpin columns (Macherey-Nagel, Düren, Germany) to remove unspent deoxynucleotide triphosphates, primers, and the short products of mis-primed PCR.
The pooled samples for low-coverage whole-genome sequencing were prepared from the population gDNA samples in equimolar concentrations. Purification was performed with the AMPure XP magnetic bead procedure (Beckman Coulter, Brea, CA, USA).
Table 1Used PCR amplicons in antibacterial TLR genes and the optimized PCR conditions.

a In reference sequences FJ147090 (TLR1), EU746465 (TLR2), AC000135.1 (TLR4), EU006635 (TLR5), and AJ618974 (TLR6).
2.2 Resequencing
The pooled amplicons from the population DNA samples were sequenced with the PacBio technology (Pacific Biosciences, Menlo Park, California, USA). The libraries for sequencing were prepared in the GATC-Biotech sequencing core laboratory (Constance, Germany) using P4-C2 chemistry according to the manufacturer's procedure. One 120 min movie was obtained using the circular consensus sequencing (CCS) protocol on the PacBio RS II machine. The primary data in the h5 format were processed into the FASTQ format with Pacific Biosciences software (SMRT Analysis Software Suite).
The library for the technology of Illumina (San Diego, CA, USA) was prepared from the pooled gDNA sample in the core laboratory of Novogene (London, UK). Two rounds of pair-end 2×150 sequencing using the X-ten technology provided 60× coverage in total. After primary processing, the resulting files in the FASTQ format were used for variant identification.
2.3 Data processing
The FASTQ files containing reads obtained with the two technologies from all three populations were mapped to the reference sequences for all five genes. The reference sequences were FJ147090 for TLR1, EU746465 for TLR2, AC000135.1 for TLR4, EU006635 for TLR5, and AJ618974 for TLR6 (White et al., 2003; Seabury et al., 2007; Seabury and Womack, 2008). The sequences were downloaded from the nucleotide databases of the National Center for Biotechnology Information (NCBI) and European Bioinformatics Institute (EBI). The mapping algorithm Geneious Mapper implemented in the Geneious program package (Biomatters, Auckland, New Zealand) was used with the minimum mapping quality equal to 50. The alignments allowed us to detect structural variants using the Geneious algorithm at the maximum variant P=0.01 and the minimum occurrence in reads equal to 1.
The exported lists of variants in comma separated text file (csv) format were carried over to the coordinates of the UMD_3.1.1 assembly of the cattle genome. The single-nucleotide polymorphisms (SNPs) arising from the differences between the original reference sequences and the current genome assembly were added. The variants were filtered for clusters resulting from a read misalignment and were defined as more than 3 variants in a 25 nt stretch.
The filtered variants were compared between both platforms. Only the variants independently detected with both the Illumina and PacBio technologies were considered to be valid. Moreover, the consistency of the allelic frequencies based on the representation of the reads was used to distinguish the valid results. The presence of the variant in the EBI European Variation Archive (https://www.ebi.ac.uk/eva/?Home, last access: 15 July 2019) was treated as an additional criterion for SNP validation.
Haplotype structure was determined directly from the simultaneous occurrence of SNPs in the long reads provided by PacBio technology.
2.4 Population comparison
Nei's standard genetic distances (Nei, 1972) were calculated for the allelic frequencies generated by the next-generation sequencing (NGS) read representation and averaged for the two technologies. Graphic representation with un-rooted trees was generated with the neighbour joining algorithm and the FigTree program package (Rambaut and Drummond, 2010) for graph visualization.
2.5 Effect prediction
The list of the validated SNPs was submitted to the Variant Effect Predictor application (VEP) (McLaren et al., 2016) of the ENSEMBL database (https://www.ensembl.org/info/docs/tools/vep/index.html, last access: 15 July 2019). The SNPs were characterized for their potential effect on the TLR product according to the localization in the protein and the characteristics of the mutated codon. In the case of the non-synonymous mutations, functional changes were estimated by calculating the SIFT (sorting intolerant from tolerant) algorithm value (Sim et al., 2012) for the protein product. The SIFT score is the normalized probability that the amino acid change is tolerated. Substitutions with scores less than 0.05 are considered to be deleterious.
Both the reference and mutant protein sequences were sent to the SWISS-MODEL server of the University of Basel (https://swissmodel.expasy.org/, last access: 15 July 2019) for homology modelling of secondary and tertiary structure. The atom coordinates in the pdb format were visualized in the graphic system Yasara (Yasara Biosciences, Vienna, Austria). The graphic models were used to localize variable amino acids on the concave or convex side of the extracellular parts of the toll-like receptors and with respect to the functional domains involved in recognition, namely leucine-rich repeats (LRRs).
3.1 Diversity
The variants found in the antibacterial TLR genes were mutually validated by their presence and frequencies in both datasets obtained with different sequencing technologies (Table 2; Novák, 2019). A total of 125 single-nucleotide change variants were detected in five genes, with 29 (23.2 %) of the variants being classified as previously unknown.
Table 2Classes of TLR-gene variants found in the Czech Red Pied and Czech Red cattle populations.
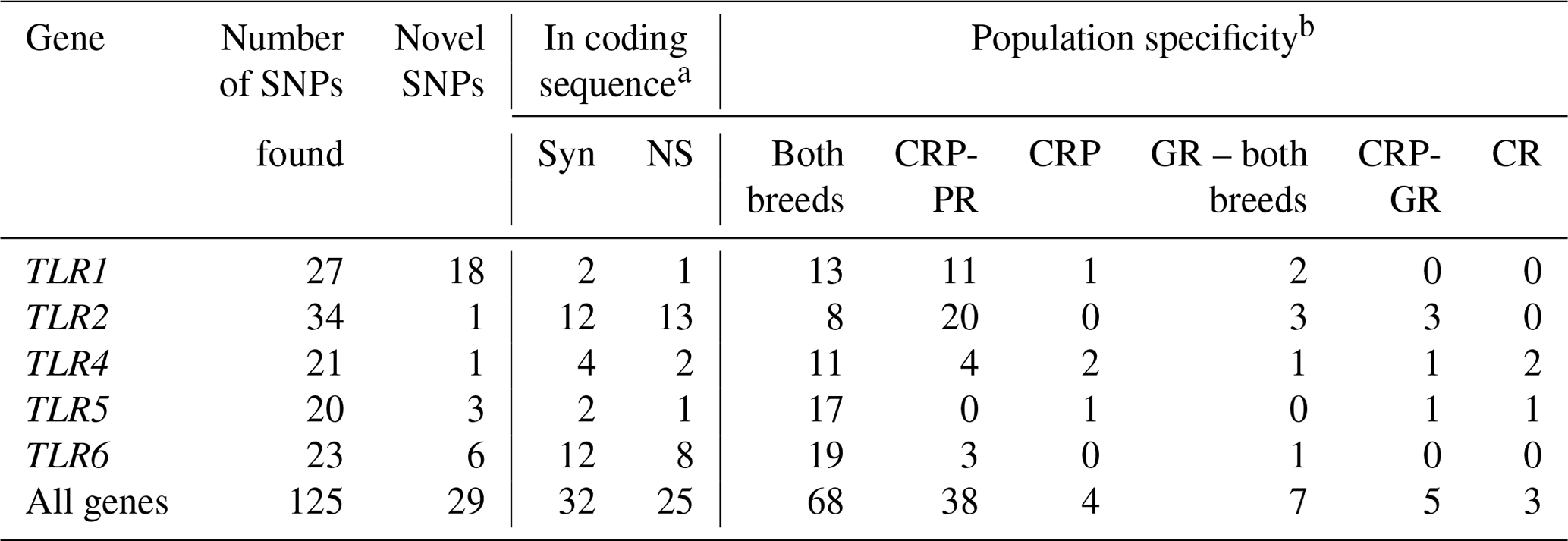
a Syn – synonymous, NS – non-synonymous. b CRP – Czech Red Pied cattle, CRP-PR – production population of CRP, CRP-GR – conserved populations of CRP in genetic resources, CR – conserved Czech Red cattle.
The number of variants found in each population can be used to characterize their total diversity: 110 in the production population of CRP, 84 in the conserved CRP subpopulation, and 78 in the Czech Red cattle. Consequently, the diversity of the conserved populations of CRP and CR was close to the diversity of the far more abundant population of current CRP, representing 76.4 % and 70.9 % of its diversity, respectively.
All three populations – the production population of CRP, and the conserved populations of CRP and CR – shared 68 (54.4 %) polymorphisms (Table 2). Thirty-eight private SNPs (30.4 %) were confined to the production population of CRP; four (3.2 %) were characteristic of CRP in a broad sense, while only five (4.0 %) were associated strictly with the conserved subpopulation of this breed. Surprisingly, only three SNPs (2.4 %) were characteristic of the ancient Czech Red breed. The CR-specific variants comprised 1414C > T (intronic) and 8218A > C (missense) in TLR4 and 25056C > T (downstream localization) in TLR5. Additionally, seven polymorphisms (5.6 %) were shared by both the conserved populations. The percentages of variants according to their population specificity, along with the individual genes, are shown in Fig. 1.
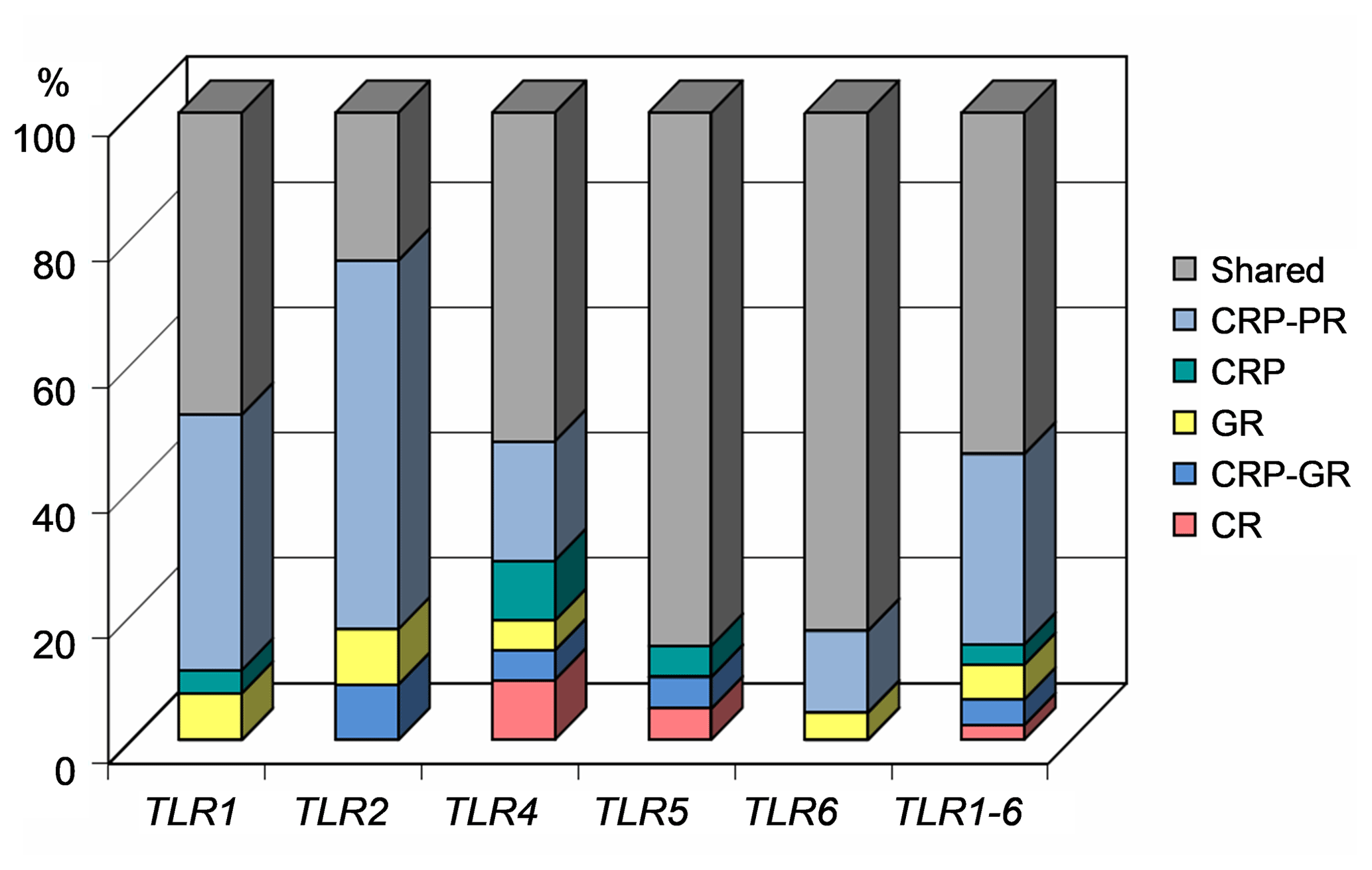
Figure 1Proportion of gene variants according to the assignment to the studied cattle populations. CRP – Czech Red Pied cattle, CRP-PR – production population of CRP, GR – conserved populations in genetic resources, CRP-GR – conserved subpopulation of CRP, and CR – conserved Czech Red cattle.
The allelic frequency derived from the read representation allowed for comparison of the population structure. The frequencies obtained with the two technologies were consistent in most of the SNPs detected and the mean value was used for subsequent interpretation. The inter-population distances based on all five TLR genes according to Nei's standard distance measure (Nei, 1972) between CRP, its conserved herd, and CR are visualized in Fig. 2.
3.2 Prediction of functional effects
The identified SNPs with breeding potential, i.e. the SNPs located in the coding sequence (CDS) or the promoter region, are presented in Table 3 along with predictions of their effects and their distribution among the populations.
In total, 57 of the found variants were located in the coding sequences, 32 of them being synonymous, and 25 non-synonymous. The ratio of non-synonymous mutations to the total number of CDS variants was 43.9 % (25 of 57) for all five genes, with the highest value, 52.0 %, in TLR2. In TLR1, intronic variants prevailed; therefore, only one variant with a moderate effect was reported by the VEP and SIFT programs and was associated with the conserved populations. Nevertheless, two intronic variants, not presented in Table 3, with a previously reported association with brucellosis (Prakash et al., 2014) were observed in all three populations. Their frequencies ranged from 0.054 to 0.143 and from 0.500 to 0.646 for SNPs 1575G > A (c.1380) and 1641A > C (c.1446), respectively.
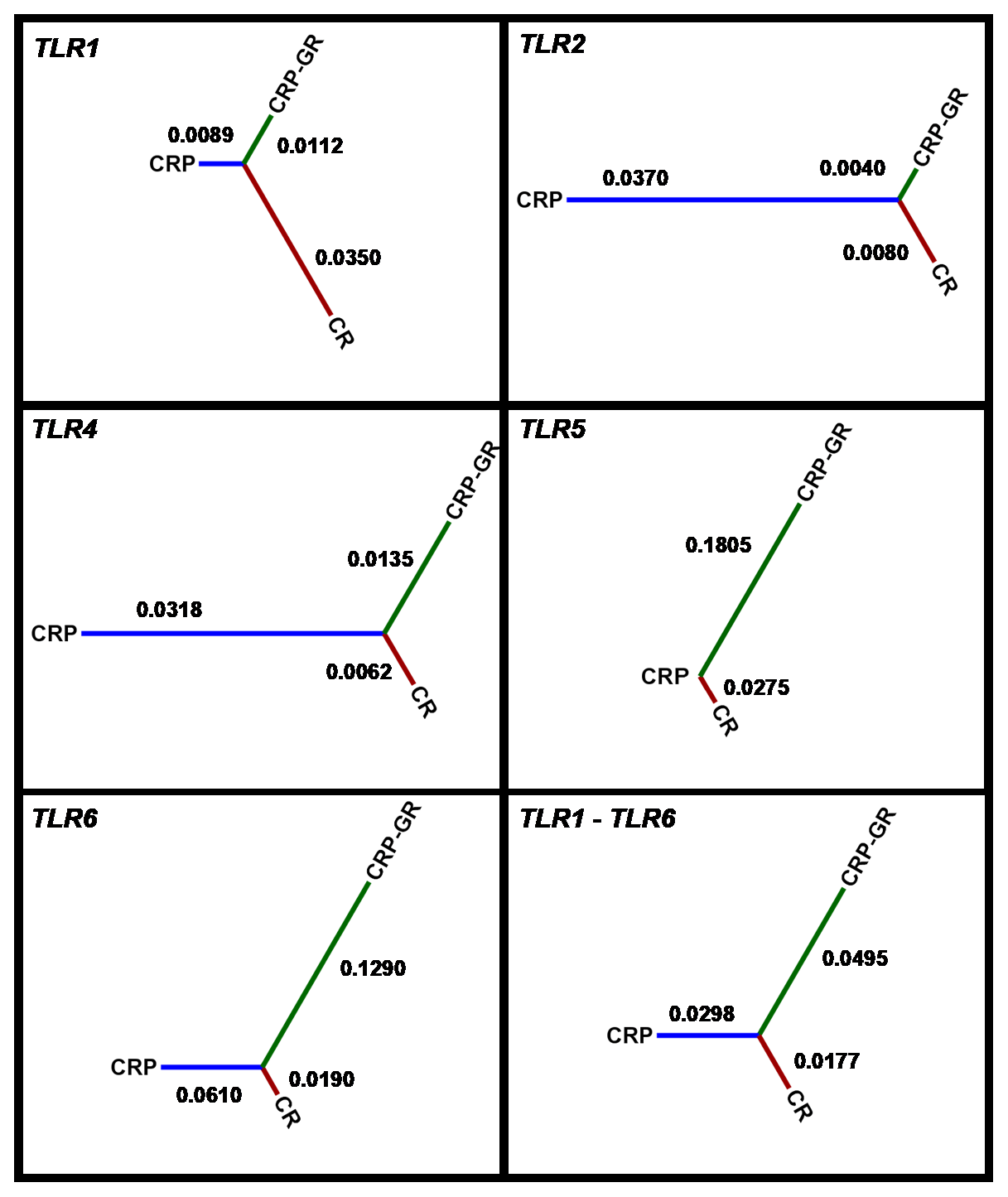
Figure 2Inter-population distances based on all five TLR genes according to Nei's standard distance measure. CRP – production population of Czech Red Pied cattle, CRP-GR – conserved subpopulation of Czech Red Pied in genetic resources, and CR – conserved Czech Red cattle.
Table 3SNPs located to the coding sequence or to the promoter region of antibacterial TLRs present in the populations of Czech Red Pied and Czech Red cattle.
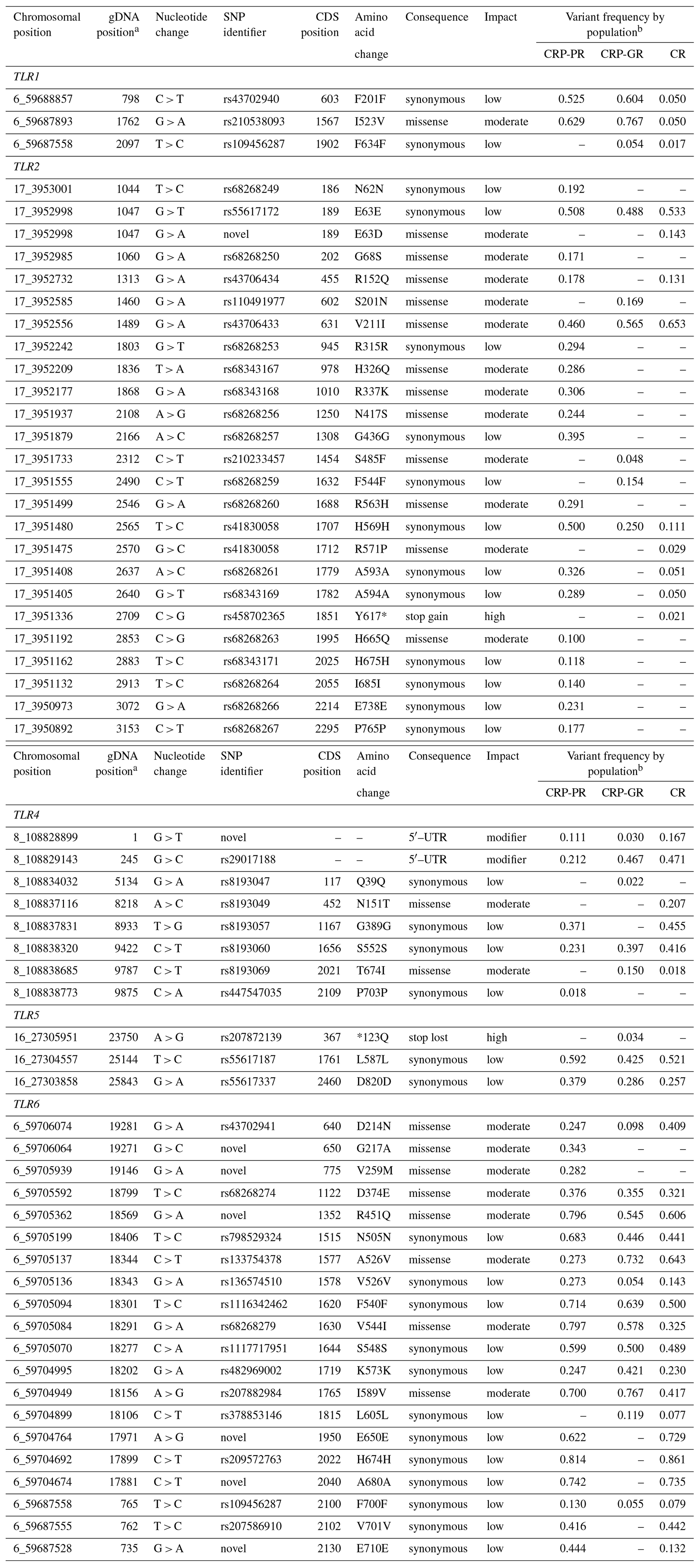
a Positions of SNPs are determined in the reference sequences FJ147090, EU746465, and AC000135.1 for TLR1, TLR2, and TLR4, respectively. In TLR5 and TLR6, the positions are relative to gene starts in UMD_3.1.1. b CRP-PR – production population of Czech Red Pied cattle, CRP-GR – conserved populations of CRP in genetic resources, and CR – conserved Czech Red cattle.
Most of the non-synonymous nucleotide changes, which are potentially important for breeding, were concentrated in TLR2 and TLR6. Of 13 missense TLR2 variants, 12 were predicted to exert a moderate effect, and only one change with a predicted high effect (Y617*) was found in the conserved Czech Red cattle.
In variants of TLR4 (Table 3), four changes with low effect and four changes with moderate effect were found, of which T674I was associated with conserved populations and N151T with CR. Four intronic variants with known functional impacts (Ruiz-Larrañaga et al., 2011) are not listed among the CDS variants in Table 3. They were 5087A > G (rs8193046), 7999A > G, c.94–24A > G (rs8193046), and c.261–28G > A (rs43578100) with frequencies ranging from 0.246 to 0.553 in all three populations. Another active intronic variant, c.94–867A > G (rs43578097), was found only in the CRP production herd with a frequency of 0.182.
In spite of considerable total variation in the TLR5 gene, only two mutations were associated with a small predicted effect and one with a large predicted impact; however, the stop-loss change *123Q (rs207872139), with a high expected impact, is confined to the conserved CRP population.
In contrast to TLR5, variability in the TLR6 gene yielded 12 synonymous SNPs with a predicted low effect and eight amino acid substitutions associated with a moderate effect. Only one of the synonymous mutations, rs378853146, was restricted to the conserved populations.
The SIFT value, which quantifies the expected impact, dropped to 0.2 or below in four amino acid changes in TLR2, namely E63D (SIFT value 0.10), R571P (<0.01), R563H (<0.01), and Y617* (<0.01), and in the R451Q (0.20) substitution in TLR6; however, even the additional 21 variants in the studied gene set characterized as causing changes with moderate impact should be considered innate immunity factors. In addition, the change *123Q in TLR5 was classified as having a high impact by the VEP program alone.
Summing up, only 5 of 45 variants that were predicted to have effects on the toll-like receptors function were restricted to the conserved populations.
4.1 TLR-gene variation in the studied herds
The study demonstrated considerable variation in the five members of the TLR family with antibacterial function in the current Czech Simmental population (Czech Red Pied cattle), in its conserved subpopulation representing the original gene pool, and in the presumably related historical breed Czech Red cattle.
Consistency with available data on the general diversity in cattle in the EBI variant database demonstrates the reliability of the hybrid sequencing (Koren et al., 2012). In this scheme, two different NGS technologies are combined to identify and eliminate the systemic faults of each. The technique of hybrid sequencing was originally suggested for obtaining correct de novo assemblies (Koren et al., 2012). This approach allows for avoidance of disambiguities originating from duplicates and large rearrangements. The principle is applicable to the validation of discovered SNPs, helping to avoid single-purpose genotyping reactions, a costly and laborious step in the discovery of polymorphisms. The application of this approach on a small scale for the description of variability in individual genes is well justified.
Nevertheless, the comparatively high proportion of novel SNPs observed (23.2 %) compared with the limited number of novel SNPs reported in an analogous study (Bilgen et al., 2016) for the Holstein population (only 4 novel SNPs of 274 for three TLR genes and 3 of 45 for TLR2) was surprising but can be ascribed to the attention focused on the Holstein population.
It should be noted that many of the observed polymorphisms have been previously reported, mostly for meat breeds, in the panel of 26 world breeds, namely 798C > T and 1762G > A in TLR1, 513C > T, 1047G > T, 1313G > A, and 2565T > C in TLR2, and 855G > A in TLR6 (Seabury et al., 2007; Seabury and Womack, 2008; Seabury et al., 2010). The polymorphisms 9787C > T in TLR4 and 855G > A in TLR6 were represented in a panel of 16 European breeds at frequencies of 0.12 and 0.52, respectively (Mariotti et al., 2009).
Application of the PacBio technology provides in addition long reads (up to 1200 bp in the present work) that allow direct phasing of located SNPs. The data obtained in the current work are thus expected to contribute to the precision of the previously studied haplotype structure of the TLR genes (White et al., 2003; Seabury et al., 2010; Ruiz-Larrañaga et al., 2011; Bilgen et al., 2016; Novák et al., 2017). This approach preliminarily revealed as many as 15 haplotypes in amplicon T2_1 of TLR2 in the studied populations, which is consistent with earlier reports of high haplotype numbers at this locus (Seabury et al., 2010; Bilgen et al., 2016).
4.2 Distinctness of conserved populations
Inclusion of the conserved subpopulation of the Czech Red Pied cattle, reflecting the genetic structure before 2000, facilitated the detection of selection trends in CRP. The diversity found in the conserved subpopulation of CRP, representing 75.4 % of the diversity of the main production population, demonstrates that gene erosion due to intensive breeding for the production traits has not occurred over the last 2 decades.
Significantly, only five SNPs were specific for the conserved historical population compared with 38 SNPs that were specific for the modern population. The low diversity of the conserved CRP population may have reflected the small number of sampled animals (n=30). In addition, the total size of the conserved CRP, which is approximately 70 animals, is close to the minimum effective population. The plausible conclusion is that after opening the breeding population to imported animals of the Fleckvieh and Montbélliard breeds, the consequent enrichment of the local Simmental population led to an increase in the available diversity in innate immunity genes, regardless of preferential breeding for utilitarian traits.
Notably, the allelic richness of the historical population of the Czech Red cattle after two historical bottlenecks remained comparable with the richness of the large population of CRP; however, the distinctness of the CR breed was characterized by only three breed-specific alleles when compared to both Simmental populations. This indicates that breed-specific diversity due to the ascribed ancient origin of the breed is not visible at the level of TLR-gene variants; moreover, the source of the breed-specific alleles might be admixture from the group of breeds that were used in the process of breed revitalization after a population bottleneck in 1987. This group of breeds included, in addition to Czech Simmental, Polish Red, German Mountain Red, Angler, and Ayrshire cattle. Full reconstruction of the specific admixture is possible on the basis of haplotype assignment (Wang et al., 2017).
The average allelic frequency-based differences among the studied populations were moderate, as characterized by Nei's distances. They corresponded to the previously demonstrated relatedness of Czech Red Pied and Czech Red cattle, as estimated using microsatellite polymorphisms in the context of other central European breeds (Čítek et al., 2006; Zaton-Dobrowolska et al., 2007).
The pattern of inter-population distances for different TLRs was consistent only between TLR2 and TLR4. On the other hand, similarity was not observed between the polymorphisms of TLR1 and TLR6, in spite of their identical localization on chromosome 6. The variation in distances in different TLRs must be partially assigned to the low number of SNPs available at individual loci.
The calculated inter-population distances did not support the distinctness of the historical breed CR, consistently with private allele occurrence. It should be noted that inter-varietal differences in bovine TLR polymorphisms are not generally high. For example, specialized beef and dairy breeds could not be differentiated despite an average polymorphism density of 1 marker per 158 bp in the TLR genes (Fisher et al., 2011).
The role of historical populations as a source of new functionally important alleles for breeding seems to be moderate; however, some of the variants restricted to the historical populations might be recruited to resistance breeding. The polymorphism H326Q in TLR2, which was identified as promising by Jann et al. (2008), was absent from the Holstein population but was observed in autochthonous Turkish breeds (Bilgen et al., 2016). In the case of CRP, this polymorphism is already present in the production herd. The 8218A > C polymorphism (N151T) restricted to CR might be a candidate for introgression after evaluation of its impact. Notably, the non-synonymous polymorphism H665Q, present in the Czech Red Pied population at 10 % frequency, is considered specific for the Bos indicus population (Jann et al., 2008).
4.3 Indications for functional consequences applicable in breeding
4.3.1 Functionally relevant polymorphism
As shown in protein modelling that included 280 variable sites in all bovine TLR genes, up to 32 % of the aa substitutions affected protein function (Fisher et al., 2011). Consequently, a comparable proportion of functional changes could be assumed for the studied populations. The observed value of 20 % (5 of 25) according to VEP and SIFT predictions of high effects is within the expected range.
4.3.2 TLR1 polymorphisms
In spite of a limited number of mutations found in TLR1 in the present work, they may contribute to resistance breeding. The non-synonymous mutation 1762G > A, present in all three populations, leads to a substitution I523V that disturbs the transmembrane region of the receptor (Russell et al., 2012). Accordingly, a higher susceptibility to bovine tuberculosis has been demonstrated for the AA and AG genotypes in Holstein cattle (Sun et al., 2012). On the other hand, the synonymous 798C > T (F201F) was tested with a negative result (Sun et al., 2012).
Synonymous 2097T > C (F634F), which was found to be limited to the conserved populations in our work, is presumably linked with 2463C > T in the 3′–UTR (untranslated region), exerting an effect on acute mastitis (Russell et al., 2012). Unfortunately, both active SNPs reported by Russell et al. (2012), namely −79T > G and 2463C > T, were out of the amplicon range and their presence was not directly observed.
In view of the epidemiological and economic importance of brucellosis, it is worth mentioning that two intronic mutations, 1575G > A (rs454341370) and 1641A > C (rs135207279), affecting this disease (Prakash et al., 2014) were present at frequencies permitting both testing in association studies and immediate use in selection. The absence of an effect on brucellosis in the otherwise active missense mutation 1762G > A (Prakash et al., 2014; Russell et al., 2012; Sun et al., 2012) suggests that the causal mutations are out of the screened range, presumably in the regulatory region of TLR1, and that linkage in the haplotype structure is critical for observation of the effect.
4.3.3 TLR2 polymorphism
Based on high diversity in TLR2 and matches with the polymorphisms in other populations, the possibility of breeding applications is higher for this gene than for other TLR genes surveyed. Jann et al. (2008) predicted the functional consequences of the TLR2 variants by defining the protein regions that show the signatures of positive selection, based on interspecific sequence comparison. The identified region corresponds to the domains participating in recognition spanning from aa 260 to 360. Of the known substitutions L227P, H305P, and H326G located in this region, H326G (rs68343167) is considered to be potentially the most efficient one because of the type of aa change; moreover, in human TLR2 this domain is crucial for ligand binding. Similarly, Bilgen et al. (2016) characterized the functional consequences of aa substitutions in TLR2 using exact localization in protein models.
Applying this information to the present purpose, most of the aa changes found in the studied populations could be placed in protein domains. E63D (in CR only) is located in LRR1, R152Q (shared by the three populations) in LRR5, I211V (shared polymorphism) in LRR7, H326G, and R563H (restricted to the CRP production herd) in LRR20, and H665Q (in the CRP production herd) is located in the highly conserved toll/interleukin-1 receptor (TIR) homology region. Positioning of these substitutions in functionally important regions increases the probability of detecting health effects in anticipated population studies. Notably, the functional effects of 2546G > A (R563H) disturbing LRR20 have been repeatedly predicted (Bilgen et al., 2016; Seabury et al., 2010; Fisher et al., 2011). The polymorphism H326G was observed in the studied populations and represents a potential target for breeding. Placement of the non-synonymous R152Q, I211V and R563H in LRRs of the extracellular part of the TLR2 protein is illustrated in Fig. 3.

Figure 3(a) Extracellular part of TLR2 with leucine-rich repeats indicated in red. (b) Placement of functionally relevant amino-acid substitutions in TLR2 found in the Czech indigenous cattle breeds.
Accordingly, the predicted effects found support on the population level. The E63D substitution in exon 2 of the TLR2 gene was reported to be associated with susceptibility or resistance to tuberculosis in cattle and was suggested for exploitation in breeding upon validation (Bhaladhare et al., 2016). On the other hand, synonymous E63E in the same protein position was uniformly distributed across all studied populations with frequencies of approximately 50 % (Table 3) facilitating potential association studies.
The change of 513C > T in the 5′–UTR increases susceptibility to paratuberculosis (PTB) (Fisher et al., 2011), while 1047G > T (E63D) and 1313G > A (R152Q) disturbing LRR1 and LRR5, respectively, increase susceptibility to bovine tuberculosis (Bilgen et al., 2016; Bhaladhare et al., 2016). The latter SNPs are components of the TLR2 haplotype that enhances resistance to PTB (Ruiz-Larrañaga et al., 2011).
Surprisingly, the C allele of the synonymous change 2565T > C (His569His) was associated with susceptibility to PTB in both the homozygous and heterozygous state (Koets et al., 2010). Accordingly, allele C is a part of the haplotype susceptible to PTB together with the neighbouring SNP rs6826825 (Phe550Phe) (Ruiz-Larrañaga et al., 2011; Koets et al., 2010). On the other hand, allele T is a part of a favourable combination of six genes for resistance to PTB (Juste et al., 2018).
Testing the complete haplotypes instead of individual polymorphisms is an approach that improves the resolution power of association studies (Ruiz-Larrañaga et al., 2011; Abdel-Shafy et al., 2014). Consequently, a risk haplotype of TLR2 for Mycobacterium avium ssp. paratuberculosis (MAP) infection CAGGCCC composed of c.455G > A, c.457T > C, c.602G > A, c.631A > G, c.1049C > T, c.1632C > T, and c.1707T > C has been identified (Ruiz-Larrañaga et al., 2011). The general character of this result is illustrated by the fact that six of these polymorphisms were also present in the studied populations (Table 3).
Besides, the ability of TLR products to form functional heterodimers (Huang et al., 2011) and to interact with other innate immunity factors should be considered in estimates of gene variants. In a recent study, Juste et al. (2018) reported combinations of polymorphisms in a set of six innate immunity genes that contributed to PTB resistance in the Holstein breed. Polymorphism R571P (rs41830058) in TLR2, a part of the most favourable polymorphism combination, was also observed in the ancient CR population.
Some of the SNPs in TLR2 were previously validated for their effects on disease resistance but were absent in the studied populations; therefore, their potential cannot be used in the current breeding scheme. They include the SNP resulting in I680V in the conserved TIR region that is associated with the MAP resistance (Mucha et al., 2009), c.1903T > C in which the C allele is associated with MAP resistance (Koets et al., 2010), and T385G, which is reported to affect mastitis incidence in Holstein, Simmental, and Sanhe cattle (Zhang et al., 2009).
4.3.4 TLR4 polymorphism
A functionally important SNP detected in TLR4 of the studied populations, c.-226G > C (rs29017188), is localized in the 5′–UTR region and is supposed to alter TLR4 expression (Sharma et al., 2008). This change may affect the binding of corresponding transcription factors c-Ets-1, MZF1, and ADR1 (Sharma et al., 2008). The susceptibility to MAP infection conferred by the C allele (Ruiz-Larrañaga et al., 2011) raises the possibility that this mutation can be exploited in breeding.
The change 9787C > T (c.2021, T674I), which is located in the transmembrane region of the receptor, is well known for its effect on susceptibility to PTB (Fisher et al., 2011) and its positive effect on milk protein and fat contents (Beecher et al., 2010); however, the CRP population was found to be already monomorphic for the favourable C allele, although variation still persists.
The high frequencies that were discovered in five functionally relevant intronic variants of TLR4 (Ruiz-Larrañaga et al., 2011) may be advantageous for phenotypic evaluation of these variants and potential use in selection. The effect of the intronic variant 5087A > G (rs8193046) with respect to PTB has been independently observed by Kumar et al. (2019) in a multi-breed population. In parallel, this effect has been demonstrated for the synonymous mutation 9422C > T (rs8193060), also found in the present study at convenient frequencies ranging from 0.231 to 0.416.
The effects of the mutation 5087A > G seem to be multilateral since an association with keratoconjunctivitis and calving ease has been reported (Mullen et al., 2018). This observation is not surprising in view of the range of known non-immune functions of toll-like receptors (Anthoney et al., 2018). In the particular case of calving ease, the effect of TLR4 variation might reflect a role in the inflammatory response and myometrial signalling before parturition, which is mediated by the MyD88/TRAF6/NF-κB pathway (Lim et al., 2017). The intronic localization of the SNP 5087A > G together with its multi-faceted effects points again to the importance of linkage in the haplotype structure for the detection of phenotypic associations.
Unfortunately, the TLR4 alleles reported to be associated with the resistance to brucellosis (c.10C > T and c.399C > T) were not identified in the studied populations.
4.3.5 TLR5 polymorphism
The aa polymorphisms R262H and F643L in TLR5, previously associated with a predicted functional impact (Fisher et al., 2011), were not found in our populations; however, the R125* mutation, reported as a candidate polymorphism for MAP resistance (Fisher et al., 2011), may be identical to *123Q (rs207872139) observed in our study. This was interpreted by the SIFT algorithm as a stop-loss mutation with significant functional consequences.
4.3.6 TLR6 polymorphism
Notably, mutation 855G > A (D214N) in TLR6, reported to increase the susceptibility to PTB (Fisher et al., 2011), was present in all populations at frequencies ranging from 0.098 to 0.409. On the other hand, none of a further three polymorphisms with ascribed functional changes, namely L43R, R87G, and F494I (Seabury et al., 2010; Fisher et al., 2011), were identified. Nevertheless, an additional seven aa substitutions with predicted moderate effects were found in TLR6 in the studied populations and are worth including in association studies.
Hybrid sequencing of population samples combining long-range amplicon sequencing with PacBio technology and direct gDNA sequencing with Illumina HiSeq technology is a fast and reliable approach to a survey of total variation in target genes. Using this approach, the scope of variants found in a series of antibacterial TLR genes in Czech Simmental cattle and related populations demonstrates sufficient diversity in the production population and rejects the possibility of negative impacts from preferential breeding for production traits. The potential contribution of the conserved genetic resources to future resistance breeding is rather moderate in this particular case and it is restricted to the non-overlapping parts of diversity. An open question is whether this observation is transferable to other rare historical breeds of cattle. The located non-synonymous single-nucleotide polymorphisms with predicted and/or previously reported effects on the innate immunity system are expected to be used as genotyping targets in association studies and breeding projects aimed at the Simmental-type breeds.
The complete list of identified SNPs has been deposited in the Open Science Framework data repository (https://osf.io/f7r4s/, Novák, 2019). Additional data are available upon request to the corresponding author.
All the authors contributed in equal measure to this work.
The authors declare that they have no conflict of interest.
The authors thank the company CHD Impuls for making archived insemination doses available for the purposes of this study.
The Long-Term Concept of the Research Institution Development of the Institute of Animal Science funded by the Ministry of Agriculture of the Czech Republic and the National Agency for Agricultural Research, project no. QJ1610489, supported this work.
This paper was edited by Steffen Maak and reviewed by two anonymous referees.
Abdel-Shafy, H., Bortfeldt, R. H., Tetens, J., and Brockmann, G. A.: Single nucleotide polymorphism and haplotype effects associated with somatic cell score in German Holstein cattle, Genet. Sel. Evol., 46, 35, https://doi.org/10.1186/1297-9686-46-35, 2014.
Anthoney, N., Foldi, I., and Hidalgo, A.: Toll and Toll-like receptor signalling in development, Development, 145, UNSP dev156018, https://doi.org/10.1242/dev.156018, 2018.
Beecher, C., Daly, M., Childs, S., Berry, D. P., Magee, D. A., McCarthy, T. V., and Giblin, L.: Polymorphisms in bovine immune genes and their associations with somatic cell count and milk production in dairy cattle, BMC Genet., 11, 99, https://doi.org/10.1186/1471-2156-11-99, 2010.
Bhaladhare, A., Sharma, D., Kumar, A., Sonwane, A., Chauhan, A., Singh, R., Kumar, P., Yadav, R., Baqir, M., Bhushan, B., and Prakash, O.: Single nucleotide polymorphisms in toll-like receptor genes and case-control association studies with bovine tuberculosis, Vet. World, 9, 458–464, 2016.
Bilgen, N., Kul, B. C., Offord, V., Werling, D., and Ertugrul, O.: Determination of genetic variations of Toll-like receptor (TLR) 2, 4, and 6 with next-generation sequencing in native cattle breeds of Anatolia and Holstein Friesian, Diversity, 8, 23, https://doi.org/10.3390/d8040023, 2016.
Boichard, D., Ducrocq, V., and Fritz, S.: Sustainable dairy cattle selection in the genomic era, J. Anim. Breeding Genet., 132, 135–143, 2015.
Čítek, J., Panicke, L., Řehout, V., and Procházková, H.: Study of genetic distances between cattle breeds of Central Europe, Czech J. Anim. Sci., 51, 429–436, 2006.
FAO.: Global Plan of Action for Animal Genetic Resources and the Interlaken Declaration, FAO, Rome, 2007.
Fisher, C. A., Bhattarai, E. K., Osterstock, J. B., Dowd, S. E., Seabury, P. M., Vikram, M., Whitlock, R. H., Schukken, Y. H., Schnabel, R. D., Taylor, J. F., Womack, J. E., and Seabury, C. M.: Evolution of the bovine TLR gene family and member associations with Mycobacterium avium subspecies paratuberculosis infection, Plos One, 6, e27744, https://doi.org/10.1371/journal.pone.0027744, 2011.
Hořín, P., Vojtíšek, P., Vyskočil, M., and Majzlík, I.: Major histocompatibility complex class I (BoLA-A) polymorphism in Czech red cattle, Czech J. Anim. Sci. (Živočišná Výroba), 42, 533–538, 1997.
Huang, Y. H., Temperley, N. D., Ren, L. M., Smith, J., Li, N., and Burt, D. W.: Molecular evolution of the vertebrate TLR1 gene family – a complex history of gene duplication, gene conversion, positive selection and co-evolution, BMC Evol. Biol., 11, 149, https://doi.org/10.1186/1471-2148-11-149, 2011.
Jann, O. C., Werling, D., Chang, J. S., Haig, D., and Glass, E. J.: Molecular evolution of bovine Toll-like receptor 2 suggests substitutions of functional relevance, BMC Evol. Biol., 8, 288, https://doi.org/10.1186/1471-2148-8-288, 2008.
Jungi, T. W., Farhat, K., Burgener, I. A., and Werling, D.: Toll-like receptors in domestic animals, Cell Tissue Res., 343, 107–120, 2011.
Juste, R. A., Vazquez, P., Ruiz-Larrañaga, O., Iriondo, M., Manzano, C., Agirre, M., Estonba, A., Geijo, M. V., Molina, E., Sevilla, I. A., Alonso-Hearn, M., Gomez, N., Perez, V., Cortes, A., and Garrido, J. M.: Association between combinations of genetic polymorphisms and epidemiopathogenic forms of bovine paratuberculosis, Heliyon, 4, e00535, https://doi.org/10.1016/j.heliyon.2018.e00535, 2018.
Kawai, T. and Akira, S.: The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors, Nature Immunol., 11, 373–384, 2010.
Koets, A., Santema, W., Mertens, H., Oostenrijk, D., Keestra, M., Overdijk, M., Labouriau, R., Franken, P., Frijters, A., Nielen, M., and Rutten, V.: Susceptibility to paratuberculosis infection in cattle is associated with single nucleotide polymorphisms in Toll-like receptor 2 which modulate immune responses against Mycobacterium avium subspecies paratuberculosis, Prev. Vet. Med. 93, 305–315, 2010.
Koren, S., Schatz, M. C., Walenz, B. P., Martin, J., Howard, J. T., Ganapathy, G., Wang, Z., Rasko, D. A., McCombie, W. R., Jarvis, E. D., and Phillippy, A. M.: Hybrid error correction and de novo assembly of single-molecule sequencing reads, Nature Biotech., 30, 693–700, 2012.
Kumar, S., Kumar, S., Singh, R. V., Chauhan, A., Kumar, A., Sulabh, S., Bharati, J., and Singh, S. V.: Genetic association of polymorphisms in bovine TLR2 and TLR4 genes with Mycobacterium avium subspecies paratuberculosis infection in Indian cattle population, Vet. Res. Commun., 43, 105–114, 2019.
Lim, R., Barker, G., and Lappas, M.: TLR2, TLR3 and TLR5 regulation of pro-inflammatory and pro-labour mediators in human primary myometrial cells, J. Reprod. Immunol., 122, 28–36, 2017.
Ludwig, A., Lieckfeldt, D., Hesse, U. G. W., and Froelich, K.: Tracing the maternal roots of the domestic Red Mountain Cattle, Mitochondrial DNA Part A, 27, 1080–1083, 2016.
Mariotti, M., Williams, J. L., Dunner, S., Valentini, A., and Pariset, L.: Polymorphisms within the Toll-like receptor (TLR)−2, −4, and −6 genes in cattle, Diversity, 1, 7–18, 2009.
Mátlová, V.: Management of farm animal genetic resources in the Czech Republic, Slovak J. Anim. Sci., 46, 127–130, 2013.
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R. S., Thormann, A., Flicek, P., and Cunningham, F.: The Ensembl Variant Effect Predictor, Genome Biol., 17, 122, https://doi.org/10.1186/s13059-016-0974-4, 2016.
Mucha, R., Bhide, M. R., Chakurkar, E. B., Novak, M., and Mikula, I.: Toll-like receptors TLR1, TLR2 and TLR4 gene mutations and natural resistance to Mycobacterium avium ssp. paratuberculosis infection in cattle, Vet. Immunol. Immunopathol., 128, 381–388, 2009.
Mullen, M. P., McClure, M. C., and Kearney, J. F.: Relationships between a TLR4 allele associated with IBK and production traits in dairy cattle, in: Book of Abstracts of the 69th Annual Meeting of the European Federation of Animal Science, Dubrovnik, 27–31 August 2018, 66.38, 2018.
Nei, M.: Genetic distance between populations, Am. Nat., 106, 283–292, 1972.
Novák, K.: Czech Red Pied TLR SNPs, full list, OSF, available at: https://osf.io/f7r4s/, last access: 17 July 2019.
Novák, K., Pikousová, J., Czerneková, V., and Mátlová, V.: Diversity of the TLR4 immunity receptor in Czech native cattle breeds revealed using the Pacific Biosciences sequencing platform, Anim. Biotech. 28, 228–236, 2017.
Prakash, O., Kumar, A., Sonwane, A., Rathore, R., Singh, R. V., Chauhan, A., Kumar, P., Renjith, R., Yadav, R., Bhaladhare, A., Baqir, M., and Sharma, D.: Polymorphism of cytokine and innate immunity genes associated with bovine brucellosis in cattle, Mol. Biol. Rep. 41, 2815–2825, 2014.
Purse, B. V., Mellor, P. S., Rogers, D. J., Samuel, A. R., Mertens, P. P. C., and Baylis, M.: Climate change and the recent emergence of bluetongue in Europe, Nature Rev. Microbiol., 3, 171–181, 2005.
Rambaut, A. and Drummond, A.: FigTree v1. 3.1, University of Edinburgh, Edinburgh, UK, 2010.
Ruiz-Larrañaga, O., Manzano, C., Iriondo, M., Garrido, J. M., Molina, E., Vazquez, P., Juste, R. A., and Estonba, A.: Genetic variation of toll-like receptor genes and infection by Mycobacterium avium ssp. paratuberculosis in Holstein-Friesian cattle, J. Dairy Sci., 94, 3635–3641, 2011.
Russell, C. D., Widdison, S., Leigh, J. A., and Coffey, T. J.: Identification of single nucleotide polymorphisms in the bovine Toll-like receptor 1 gene and association with health traits in cattle, Vet. Res., 43, 17, https://doi.org/10.1186/1297-9716-43-17, 2012.
Seabury, C. M. and Womack, J. E.: Analysis of sequence variability and protein domain architectures for bovine peptidoglycan recognition protein 1 and Toll-like receptors 2 and 6, Genomics, 92, 235–245, 2008.
Seabury, C. M., Cargill, E. J., and Womack, J. E.: Sequence variability and protein domain architectures for bovine Toll-like receptors 1, 5, and 10, Genomics, 90, 502–515, 2007.
Seabury, C. M., Seabury, P. M., Decker, J. E., Schnabel, R. D., Taylor, J. F., and Womack, J. E.: Diversity and evolution of 11 innate immune genes in Bos taurus taurus and Bos taurus indicus cattle, P. Natl. Acad. Sci. USA, 107, 151–156, 2010.
Sharma, B. S., Leyva, I., Schenkel, F., and Karrow, N. A.: Association of toll-like receptor 4 polymorphisms with somatic cell score and lactation persistency in Holstein bulls, J. Dairy Sci., 89, 3626–3635, 2006.
Sharma, B. S., Mount, J., and Karrow, N. A.: Functional characterization of a single nucleotide polymorphism in the 5'-UTR region of the bovine toll-like receptor 4 gene, in: Animal Genomics for Animal Health, edited by: Pinard, M.-H., Gay, C., Pastoret, P.-P., and Dodet, B., Book Series: Developments in Biologicals, 132, Karger Publishers, Basel, Switzerland, 331–336, 2008.
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C.: SIFT web server: predicting effects of amino acid substitutions on proteins, Nucl. Acids Res., 40, W452–W457, 2012.
Sun, L. P., Song, Y. P., Riaz, H., Yang, H. Z., Hua, G. H., Guo, A. Z., and Yang, L. G.: Polymorphisms in toll-like receptor 1 and 9 genes and their association with tuberculosis susceptibility in Chinese Holstein cattle, Vet. Immunol. Immunopathol., 147, 195–201, 2012.
Wang, Y., Segelke, D., Emmerling, R., Bennewitz, J., and Wellmann, R.: Long-term impact of optimum contribution selection strategies on local livestock breeds with historical introgression using the example of German Angler cattle, G3-Genes Genom. Genet., 7, 4009–4018, 2017.
White, S. N., Taylor, K. H., Abbey, C. A., Gill, C. A., and Womack, J. E.: Haplotype variation in bovine Toll-like receptor 4 and computational prediction of a positively selected ligand-binding domain, P. Natl. Acad. Sci. USA, 100, 10364–10369, 2003.
Zaton-Dobrowolska, M., Čítek, J., Filistowicz, A., Řehout, V., and Szulc, T.: Genetic distance between the Polish Red, Czech Red and German Red cattle estimated based on selected loci of protein coding genes and DNA microsatellite sequences, Anim. Sci. Pap. Rep., 25, 45–54, 2007.
Zhang, L. P., Gan, Q. F., Ma, T. H., Li, H. D., Wang, X. P., Li, J. Y., Gao, X., Chen, J. B., Ren, H. Y., and Xu, S. Z.: Toll-like receptor 2 gene polymorphism and its relationship with SCS in dairy cattle, Anim. Biotech., 20, 87–95, 2009.