
the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Feb 2018
| 13 Feb 2018
Invited review: Genetic and genomic mouse models for livestock research
Deike Hesse
Gudrun A. Brockmann
Knowledge about the function and functioning of single or multiple interacting genes is of the utmost significance for understanding the organism as a whole and for accurate livestock improvement through genomic selection. This includes, but is not limited to, understanding the ontogenetic and environmentally driven regulation of gene action contributing to simple and complex traits. Genetically modified mice, in which the functions of single genes are annotated; mice with reduced genetic complexity; and simplified structured populations are tools to gain fundamental knowledge of inheritance patterns and whole system genetics and genomics. In this review, we briefly describe existing mouse resources and discuss their value for fundamental and applied research in livestock.
- Article
(419 KB) - Full-text XML
- BibTeX
- EndNote





During the last 10 years, tools for genome analyses have developed tremendously. Along with the technological progress to examine whole genomes quickly and accurately, these techniques became flexible in size and origin of material to be analyzed. The wide range of use made these techniques affordable for diverse applications. In animal research, these techniques changed the accessibility of livestock genomic information dramatically. Whole genome sequence data are available for the most widely used species; these include cattle, pig, sheep, goat, buffalo, rabbit, horse, camel, and alpaca. These data are available as a reference genome sequence (Ensembl; Zerbino et al., 2017, https://www.ensembl.org/). Variations for other breeds are stored in the form of sequence variants relative to this reference sequence (dbSNP; Sherry, 2001, https://www.ncbi.nlm.nih.gov/SNP/). In addition, often detailed pedigree structures are known and diverse phenotypes are collected for selected livestock populations. For the improvement of breeds, sequence variants are associated with animal characteristics and resulting allele effects are used for selection, either single gene selection or genomic selection taking into account gene effects across the whole genome. Moreover, ingenious methods for the generation of targeted mutations found their way from model animals to livestock species. Through this progress, model organisms attain a new position in fundamental science supporting the elucidation of genetics and genomics in livestock.
Genetics and genomics are often intermingled. The reason lies in the deeper insight into the genome itself. While classical genetics is the study of the heredity of a phenotypic trait, explaining the transmission of genes together with the expected visible phenotype from parents to offspring, genomics is the study of the genome as a carrier of the genetic information of an individual in its entirety. Genomics aims to elucidate genomic structure, modulation, functioning, and interaction as a whole and their action after transmission of genome parts in reproduction. As such, genomics can be considered as a subfield or as an extension of genetics.
In this review, we revisit the benefits of mouse models for the current research in livestock genetics and genomics. We briefly describe existing mouse resources and tools for investigating simple and complex traits and discuss those resources for the generation of knowledge for fundamental and applied research in livestock, where we focus on mammalian species.
Recently, Schmid and Bennewitz (2017) have reviewed the state of genome-wide association studies (GWASs) for mapping genes and causative mutations of quantitative traits in livestock. Information from GWASs in different livestock species has successfully contributed to further genetic improvement of populations through genomic selection, in particular in dairy cattle such as the Holstein Friesian. Nevertheless, there is a strong desire to gain further insight into the genetics and genomics of complex traits in livestock to improve genomic selection accuracy, to study the function and regulation of genes, and to understand the whole genome and fine-scale genetic architecture of genome-wide determinants of complex traits throughout lifespan and under different environments (Goddard et al., 2016). In addition to direct investigation in the target livestock species, comparative studies using model organisms as well as human genetics and genomics contribute to accomplishing this task. To facilitate comparative research, the Alliance of Genome Resources provides an interface that allows users to search for and view genes, functional data, and disease associations from databases of the fly, mouse, rat, yeast, nematode, and zebra fish (http://www.alliancegenome.org, last access: 3 January 2018).
Mice are mammals, sharing 92 to 95 % of protein coding genes with humans and other mammalian livestock species, such as cattle (Elsik et al., 2009), pigs (Humphray et al., 2007), sheep (Iannuzzi et al., 1999), and goats (Schibler et al., 1998). The mouse genome is structured into 19 autosomes and the sex chromosomes. The mouse genome is around 2.7 giga base pairs long (Golden Path Length, Genome Reference Consortium mouse reference GRCm38.p5, ensemble.org, release 91, last access: 3 January 2018) and contains about 22 600 protein coding genes, 15 000 non-coding genes, and 12 000 pseudogenes, which are estimated to produce about 130 000 transcripts in total.
The mouse has always been a favorite model animal since it is small and cheap to house, and mice are highly reproductive. They can produce four to five generations per year with up to 15 animals per litter, or even more. Mice have on average a 30 times more accelerated lifespan compared to humans. Mouse embryos can easily be cryopreserved and thereby stored long term for low costs without keeping the whole colony alive and without the risk of deriving new mutations. Moreover, the genome of the mouse can be genetically modified using different techniques (for an overview see Sect. 5), which allows the study of gene functions or allele effects in the whole organism as well as in specific organs or cell types.
The biggest advantage of using mice in research is the ability to control genetics and environment. Inbred lines allow the repeated investigation of the same genotype. Genetics is controlled by targeted mating. By controlling the management conditions and housing environment, external influences can be minimized. This allows us to perform challenge or intervention studies, such as studies with pathogens, testing drug effects, diet effects, and dosage with low numbers of animals.
For the mouse, diverse inbred strains, outbred populations descending from structured crosses between inbred strains, outbred selection populations, and genetically modified animals are available. For many inbred strains, phenotypic data and gene expression data were collected and whole genome sequence data were generated and are publicly available. The wide range of strains and populations and the comprehensive collection of phenotypic and genetic data made the mouse one of the most well-studied animal models in biological research.
4.1 Classical inbred and recombinant inbred strains
A peculiarity of the mouse is the wide range of available inbred strains that were created over the past century. The advantage of these strains for genetic research is the reduction of complexity within each strain on the one hand and the maintenance of genetic diversity between strains on the other hand. Within each strain, the genome is reduced to one allele variant at each locus, while the whole set of all available inbred strains represents a wide spectrum of alternative alleles at each locus. Since all mice are genetically identical within each inbred strain, subsequent experiments addressing different questions can be performed on the same genetic background. Due to the fixation of the genotype, phenotypic variation is small within a strain and therefore the number of animals required for experiments can be minimized.
4.1.1 Classical inbred strains
In random brother–sister mating over more than 20 generations of mice of different origin led to the generation of a high variety of so-called classical inbred strains (Lyon, 1989). Currently, about 250 classical inbred strains are available at the Jackson Laboratory (https://www.jax.org/, last access: 3 January 2018). Each of these inbred strains contains a unique genome. The uniqueness of a strain depends on the evolutionary history of the founding population as well as on natural and artificial selection during the generation of the strain. The three main subspecies contributing to the different inbred strains are Mus musculus domesticus (found in western Europe, southwestern Asia, the Americas, Africa, and Oceania), Mus musculus musculus (eastern Europe and northern Asia), and Mus musculus castaneus (southern and southeastern Asia). Two additional subspecies have been recognized recently (Wilson and Reeder, 2005): Mus musculus bactrianus (central Asia) and Mus musculus gentilulus (Arabian peninsula; Madagascar). Full information on inbred mice and their origin can be found elsewhere (Brockmann, 2005).
Of the classical inbred strains, 43 were intensively phenotyped for a diverse range of traits (Brockmann and Bevova, 2002; Svenson et al., 2007). These phenotypes are available at the Mouse Phenome Database of the Jackson Laboratory (https://phenome.jax.org/, last access: 3 January 2018).
Due to genetic drift, residual unfixed alleles, spontaneous mutations, or genetic contamination from other mouse strains sub-strains from well-characterized classical inbred strains have emerged. For example, for C57BL/6, one of the most widely used classical inbred strains strain, several well-described sub-strains developed unplanned during the breeding history in different laboratories. These sub-strains do not only differ in a few mutations but also in their phenotypes (Bryant et al., 2008; Mekada et al., 2009). This has to be considered when designing experiments, but can be exploited for experiments with these strains.
For C57BL/6J, which is the mouse reference sequence at the Ensembl Genome browser (https://www.ensembl.org/, last access: 3 January 2018), and additional 35 mouse inbred strains fully annotated genome sequences are available (https://www.sanger.ac.uk/science/data/mouse-genomes-project/, last access: 3 January 2018). These strains are representative for the different genetic backgrounds occurring among laboratory mouse strains, including the three main wild subspecies. The annotated sequence information helps researchers to draw more generally applicable conclusions from experiments with these strains, which can be translated to other species.
4.1.2 Recombinant inbred strains (RISs)
Recombinant inbred strains (RISs) (Bailey, 1971) are sets of mouse strains that were derived from initial crosses between two or more inbred mouse strains and subsequent inbreeding of random pairs of siblings. Such a set of RIS represents a population that mimics simplified genetic diversity. Each single recombinant inbred strain represents one unique genotype, which is composed of randomly distributed genome parts of the founder lines. Therefore, a set of such strains can be used for association studies to finely map genetic loci responsible for phenotypic differences between the founder strains. Since every RIS is inbred, it needs to be genotyped only once, while phenotyping of the strains can be performed repeatedly. This allows a system analysis of the whole genome and multiple phenotypes simultaneously.
One of the most comprehensively investigated RIS sets is the BXD recombinant inbred strain panel comprising of 198 RIS mice (http://www.Genenetwork.org). The RISs were generated from an initial cross between C57BL/6J and DBA/2J. For C57BL/6J and DBA/2J whole genome sequence data exist and each RIS was genotyped using single-nucleotide polymorphism (SNP) chips. Therefore, every measured phenotype can be immediately associated with a causative genome region, gene or even sequence variant. In addition to association mapping of genes for single traits, these RIS resources also provide insight into the interaction between genes in the genome. It has been observed repeatedly that the phenotype of a recombinant inbred strain, as a mixture of the parental genomes, is significantly more extreme than either parental strain (e.g., Dogan et al., 2013).
The most diverse set of recombinant inbred strains so far is the Collaborative Cross (CC) (Complex Trait Consortium, 2004). The strains were constructed from a cross between eight founder mouse inbred strains (Fig. 1) (A/J, C57BL/6J, 129S1/SvImJ, NOD/ShiLtJ, NZO/HiLtJ, CAST/EiJ, PWK/PhJ, WSB/EiJ), which represent the most diverse genetic background including wildly derived mice (Complex Trait Consortium, 2004). Currently, inbreeding of around 70 CC strains has finished, with more strains to come in the near future (https://csbio.unc.edu/CCstatus/, last access: 3 January 2018). These strains can be used in the same ways as the BXD set. Since the eight parental strains capture high genetic diversity and phenotypic variation for many traits, genetic mapping and fine mapping of causal loci contributing to complex trait variation is feasible. Whole genome sequence data of all eight parental strains and imputed sequence information of the CC strains (Oreper et al., 2017) allows identification of potential functionally acting candidate genes or regulatory elements.
4.2 Outbred populations
In sharp contrast to inbred strains, outbred populations are heterogeneous stocks (HSs) that could originate from crosses of two or more inbred strains or wild mice. These populations produce high phenotypic variance since each individual carries a unique combination of the genomes of the founder strain. These populations best mimic the genetic and phenotypic heterogeneity of natural livestock populations. Nevertheless, these individuals are still simplified in their genome composition.
Through repeated random mating (or repeated selective mating for a trait in selection populations) over many generations, recombination events between founder genomes are enriched in each individual and, therefore, the genomes of the original founders are highly fragmented. The origin of each genome segment can be traced back to a founder strain and its sequence variants. Thereby, these populations facilitate fine mapping of causal genes contributing to diverse phenotypes. In addition, these populations allow for systems analysis of interaction between genome regions and interference between phenotypes as well as the search for allelic imbalances and genetic imprinting of alleles.
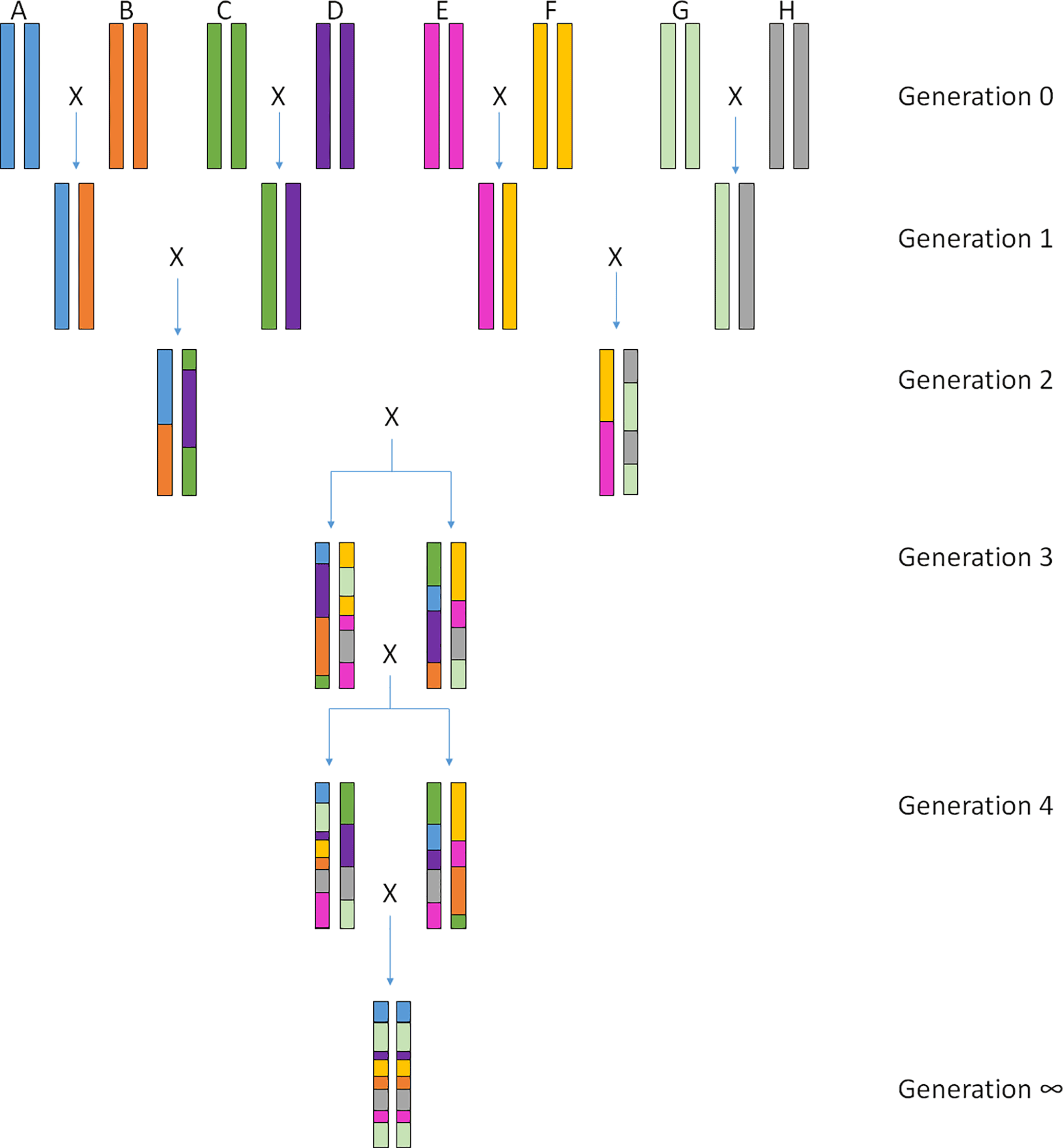
Figure 1Crossing scheme of the Collaborative Cross lines. The eight founder lines are crossed to produce generation 1 (G1) in which each mouse is heterozygous for two of the founder lines. Individuals from different G1 crosses are then mated together to produce G2. Here animals have a heterozygous genome with contributions from four of the eight founder strains. G2 individuals are then crossed with other G2 individuals who were generated using the remaining four founders. The genomes of individuals from the G3 generation are now composed of contributions from all eight founders. At this stage, siblings are mated within each of these “funnels”, leading to a recombinant inbred line by fixation of the founder alleles. After 20 generations of repeated brother–sister mating, Collaborative Cross individuals are more than 95 % homozygous across the genome. The figure was inspired by an image published in Complex Trait Consortium (2004).
4.2.1 Heterogeneous stock (HS)
HSs were created to produce genetically heterogeneous highly reproductive stocks. The following eight progenitor inbred strains were crossed to identify genes associated with addiction to alcohol and drugs: A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, CBA/J, DBA/2J, and LP/J (Hitzemann et al., 2002). Another HS population, originating from the progenitor strains C57BL/6J, BALB/cJ, RIII, AKR, DBA/2, I, A/J, and C3H was intensively investigated for genes controlling animal behavior. In this cross, the gene regulator of G-protein signaling 2 (Rgs2) was identified, which encodes a regulator of G-protein signaling. This gene modulates anxiety accounting for ∼ 5 % of the phenotypic variance in the population. The gene was first mapped in the HS population to a narrow region on chromosome 1, then remapped in the MF1 heterogeneous outbred population, and finally identified as a causal gene via genetic complementation with a Rgs2 knockout mouse (Yalcin et al., 2004).
4.2.2 Diversity outbred (DO) mice
Another heterogeneous population is the diversity outbred (DO) population, derived from the same founder strains as the CC panel of RIS (A/J, C57BL/6J, 129S1/SvImJ, NOD/ShiLtJ, NZO/HiLtJ, CAST/EiJ, PWK/PhJ, and WSB/EiJ). The biggest difference between DO and CC mice is that DO mice are not an eternal resource since they outbreed each generation. This leads to high (single gene) mapping resolution, but comes at the cost of having to genotype each individual. This loss of power is compensated for by the fact that software, such as DOQTL (Gatti et al., 2014) and R/qtl (Arends et al., 2010; Broman et al., 2003), is able to reconstruct parental haplotypes of the DO mice, allowing for more powerful haplotype association analysis.
4.2.3 Advanced intercross population (AIL)
A special case of heterogeneous stocks are advanced intercross populations (AILs). These are generated from an initial cross between two inbred strains and subsequent repeated randomized mating of members of one and the same generation to produce offspring for the next generation. An effective population size of 100 animals (50 successive mating pairs) is needed to minimize genetic drift in such a population (Darvasi and Soller, 1995). AIL mice have been very successfully used for fine mapping of causal genes. Recently, an AIL originating from a cross between C57BL/6NCrl and the Berlin Fat Mouse inbred strain was used in combination with complementation tests to identify Bbs7 as the causal gene for obesity in the Berlin Fat Mouse (Arends et al., 2016).
5.1 Tools for functional tests of genes
The function of a gene can be investigated by testing its phenotypic effect after destruction or addition to a genome by knocking it out or in, respectively. The effect can act on the whole organism, in target tissues or cell types permanently, or under specific conditions. In recent years, the gene manipulation tools have been extended by gene editing, a protocol that targets single nucleotides or short DNA sequences and allows subtle or extreme, deleterious or advantageous modifications of DNA to study such effects on genes, proteins, or regulatory elements (Gupta and Musunuru, 2014).
5.2 IMPC
The biggest consortium for the annotation of gene functions is the International Mouse Phenotyping Consortium (IMPC), an international collaboration between the European Conditional Mouse Mutagenesis Program (EUCOMM), the Knockout Mouse Project (KOMP) (USA), the North American Conditional Mouse Mutagenesis Project (NorCOMM) (Canada), and Texas A & M Institute for Genomic Medicine (TIGM) (USA). The goal of IMPC is to generate a knockout mouse for every gene in the mouse genome (Brown and Moore, 2012). IMPC-generated knockout mice are subjected to the “Adult and Embryonic Phenotype Pipeline“, which is a collection of standardized phenotyping protocols for the characterization of diverse genetically modified mouse strains. Currently, IMPC has finished the phenotyping pipeline for ∼ 4500 knocked out genes. All data generated by IMPC are publicly available online (https://www.mousephenotype.org, last access: 3 January 2018).
5.3 ENU
N-ethyl-N-nitrosourea (ENU), an alkylating agent, is one of the most effective chemicals for obtaining mutations. The mutation rate is dosage and strain dependent in male mouse sperm stem cells (Cordes, 2005). ENU produces preferentially point mutations, and these mutations occur randomly (Nolan et al., 2002). Screens for phenotypes of ENU-treated mice that deviate from normality can identify single gene effects independently of the position of the mutation in the gene. As such, ENU mutations provide a finely grained dissection of gene or protein function. This allows discovery of gene and protein functions in an unbiased way. A disadvantage is that several mutations could occur simultaneously in one individual. Therefore, several rounds of backcrossing are carried out before the phenotype screen is performed. ENU screens successfully contributed to the identification of lethal defects and genes affecting immune cell function and differentiation (Nguyen et al., 2011). Mice from ENU screens are available on request from researchers or can be purchased from the Jackson Laboratory.
5.4 Gene editing
This method can edit the DNA sequence well directed at almost every position in the genome and thereby improve or impair the gene function. This permits targeted analyses of functional domains of a gene product as well as the regulation of a gene (Esvelt et al., 2013; Gupta and Musunuru, 2014). Effective protocols that exist for gene editing use guide RNA in combination with the enzyme recombinase (CRISPR/Cas9), zinc finger nuclease, or zinc finger protein in combination with a nuclease (TALEN) (Gupta and Musunuru, 2014). Although these methods can be applied easily to livestock species, the mouse still has the unequal advantage of a short generation interval, which shortens the time for testing the effects of the introduced mutation several fold.
6.1 Key mouse models for milk and meat production, growth, and body composition
6.1.1 DGAT1
The diacylglycerol O-acyltransferase 1 (DGAT1) is a rate-limiting enzyme in the synthesis of triglycerides. Mice lacking DGAT1 can still synthesize triglycerides; however, they exhibit lactation defects because they do not produce milk (Smith et al., 2000). Knowing this function, DGAT1 was proposed as the most promising positional candidate gene for a narrow region on Bos taurus autosome (BTA) 14 with a highly significant effect on milk yield and milk fat content in a GWAS with high-performance Holstein dairy cattle (Thaller et al., 2003). This led to the identification of the DGAT1 protein mutation K232A (Grisart et al., 2002) and a promoter variant (Fürbass et al., 2006) that are segregating in Holstein but also other dairy cattle breeds and are considered causal.
6.1.2 Myostatin
Myostatin (MSTN) is one of different growth and differentiation factors (number 8, Gdf8). This factor regulates muscle cell growth in a negative feedback loop (McPherron et al., 1997). Myostatin knockout mice have approximately twice as much muscle mass as normal mice (McPherron and Lee, 1997). The phenomenon of hypermuscularity has been observed for a long time in beef cattle and sheep. In Belgian Blue cattle, the genomic locus accounting for the double muscling phenotype was finely mapped to a region containing myostatin (Grobet et al., 1997; Kambadur et al., 1997). Sequence analysis identified a 11-base pair deletion in the third exon leading to a frame shift in the coding sequence and consequently to the lack of intact myostatin (Grobet et al., 1997). Soon after discovering the myostatin effect, additional mutations were identified in the myostatin gene that are associated with increased fiber number and/or fiber diameter (McPherron and Lee, 1997) .
Double muscling was also known for Texel sheep, found on the Dutch Frisian island of Texel. Different from cattle, the availability of myostatin is here reduced to about one-third through impaired translation. A mutation in the 3' untranslated region of the gene creates binding sites for three micro RNAs of which two occur in the skeletal muscle and bind to the RNA, which interferes with the translational machinery (Clop et al., 2006).
More recently, the CRISPR/Cas9 system was employed to genetically manipulate pigs, rabbits, and goats to disable the myostatin gene with the goal of increasing meat production (Guo et al., 2016; Wang et al., 2017). In all species, the resulting animals were significantly more muscular. However, rabbits without a functional myostatin gene also exhibited signs of the enlarged tongue phenomenon, a higher rate of still births, and a reduced lifespan.
6.1.3 Leptin, its receptor, and additional adipomyokines
Leptin is a satiety hormone secreted from adipocytes and signals fat storage to the brain. Leptin as well as its receptor were identified in the obese (ob) and diabetic (db) mouse strains as natural occurring deleterious mutations in the C57BL/6J mouse inbred strain. Both the ob and the db strains are obese. The discovery of leptin and afterwards the leptin receptor through genetic mapping, fine mapping, and comparative sequencing (Chua et al., 1996; Zhang et al., 1994) was a milestone in the field of obesity research in humans and opened the doors for investigating leptin effects on energy consumption and partitioning in many species. In different livestock species, associations with many production traits, such as body composition, intramuscular fat content (Williams, 2008), feed efficiency (DeVuyst et al., 2007; Kononoff et al., 2005), reproduction (Agarwal et al., 2009), and milk performance were found (Giblin et al., 2010). Animals with high levels of circulating leptin tend to exhibit a higher feed efficiency (6.4 % feed efficiency) compared to animals with low levels of circulating leptin (8.2 % feed efficiency) (DeVuyst et al., 2007; Kononoff et al., 2005).
A recent cross-species approach using mice to model cattle identified novel putative adipomyokines involved in the cross talk between muscle and adipose tissue (Schering et al., 2015). The researchers identified 119 myokines, 79 adipokines, and 22 adipomyokines, which were subjected to network analysis to reveal remodeling of the extracellular matrix and tissue fibrosis as relevant annotation of these candidate adipomyokines. These novel adipomyokines could be interesting targets since they show a physiological role in exercise adaptation and meat quality of farm animals.
6.1.4 Gut microbiota
Germ-free mice as well as the huge diversity across inbred mouse strains provide good models to study the effect of the microbiome on disease development and resistance (Flowers and Ellingrod, 2015; Kinross et al., 2011; Rosshart et al., 2017) and to study the host–microbiota interaction (Al-Asmakh and Zadjali, 2015). Recent studies showed, for example, that germ-free mice are resistant to obesity when consuming a high-fat, high-carbohydrate Western diet (Rabot et al., 2010). Even if the effects may be inflated because of a lack of natural immune system in these mice, gnotobiotic mouse models are very useful to dissect mechanisms underlying microbe–host interactions (Clavel et al., 2016). These insights improve our understanding and provide input into investigations on livestock species and how microbiota influence not only the digestion of food but also how they affect the body's metabolism, behavior, and health.
6.2 Mouse as a model for animal diseases
6.2.1 Trypanosomiasis
Trypanosomiasis affects vertebrate livestock species in South America, northern Africa, and the Middle East. Affected species include horses, donkeys, mules, cattle, buffalo, deer, camels, llamas, dogs, and cats (Rjeibi et al., 2015). Known transmission vectors of the parasite include horseflies and vampire bats. Mouse models have been developed to study the infection routes and pathogenicity of Trypanosoma, reviewed by Antoine-Moussiaux et al. (2008), Blom-Potar et al. (2010), and Síma et al. (2011). Mouse models have played an essential role in finding genetic loci underlying resistance or susceptibility to trypanosomiasis (Foote et al., 2005). The availability of high-density linkage maps, the genome sequence, and transcriptomic tools have made the mouse essential to studying these fundamental aspects of the host–parasite interaction.
6.2.2 Bovine spongiform encephalopathy (BSE)
Bovine spongiform encephalopathy (BSE), a prion-caused disease, is a fatal and progressive degenerative disorder of the central nervous system of cattle, which can be transmitted to humans. In 1997, it was shown that transmission of BSE to transgenic mice was possible (Scott et al., 1997), and in 2006, transmission of BSE to wild-type mice was shown to be possible (Baron et al., 2006). Multiple mouse models have been developed to study prion diseases with different susceptibilities for bovine, sheep, mink, and porcine spongiform encephalopathy (Baron et al., 2007; Wilson et al., 2012). Additionally, fundamental research into prion diseases in mice (Telling, 2011) has contributed significantly to our understanding of the molecular mechanism and structure of prions as well as disease progression and transmission (Riek et al., 1996).
6.2.3 Foot-and-mouth disease
Although mice do not have hooves similar to cattle, and many other differences exist between mice and ruminant immune responses, a mouse model was developed for studying food-and-mouth disease (FMD) (Skinner, 1951). Intraperitoneal infection of unweaned mice from the FMD virus (FMDV) leads to fatal symptoms characterized by muscular paralysis (of the hind legs), degenerative changes in the myocardium and skeletal muscles, and respiratory distress within 24 h of infection, whereas infection in older mice (after weaning) is typically subclinical (Skinner, 1951). The suckling mouse model is considered a major milestone in FMDV research. Following intraperitoneal challenge, the virus replicates primarily in the pancreas and the viremic period is short (Charleston et al., 2011). FMD pathogenesis in adult mice is dependent on the genetic background of the mouse strain (with C57BL/6 mice being most susceptible), strain of virus, and challenge method. The mouse model provides a platform to evaluate therapeutics and vaccine candidates at a reduced cost and allows access to genetic mutations not available for target species, providing a powerful and versatile experimental system to interrogate the immune response to FMDV (Habiela et al., 2014).
6.2.4 African swine fever virus (ASFV)
The virus causes a haemorrhagic fever with high mortality rates in pigs, but it is also found to infect wild hosts such as warthogs, bush pigs, and ticks (Tulman et al., 2009). In 1994, under laboratory conditions, a mouse model was developed with severe combined immunodeficiency (SCID) to investigate ASFV. Injection of swine peripheral blood mononuclear cells into SCID mice results in a long-term establishment of a functional swine immune system in these mice (Revilla et al., 1994). Mouse models have also been used to test the different swine fever proteins for the immunogenicity and safety with the goal to develop vaccines for ASFV (Chen et al., 2016).
7.1 Mouse Genome Informatics (MGI)
The Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org/, last access: 3 January 2018) maintained by the Jackson Laboratory (Bar Harbor, Maine, USA) provides the most comprehensive information about the characteristics of mouse strains. Information inside this database includes the following.
-
Gene-based information. This includes basic information concerning gene location, family, ontology, and functional annotation.
-
Phenotype information and mutant alleles. This includes all information regarding spontaneous, induced, and genetically engineered mutations and their strain-specific phenotypes.
-
Human–mouse disease connection. This part was designed to facilitate the identification of published and potential mouse models of human disease, the discovery of candidate genes, and the investigation of phenotypic similarity between mouse models and human patients.
-
Recombinase (Cre) activity. MGI collects and annotates expression and activity data for recombinase-containing transgenes and knock-in alleles.
-
Functional annotation using the gene ontology. MGI's Gene Ontology project provides functional annotations for mouse gene products using the gene ontology (Ashburner et al., 2000; Gene Ontology Consortium, 2017).
-
Strains, SNPs, and polymorphisms. MGI integrates comparative data on inbred strain characteristics including SNPs, polymorphisms, and quantitative phenotypes.
-
Vertebrate homology. MGI contains homology information for mouse, human, rat, chimp, and dog.
-
Biochemical pathways (MouseCyc). This includes a database of curated biochemical pathway data for the laboratory mouse that can be integrated with functional and phenotypic data from MGI (Evsikov et al., 2009).
7.2 Mouse Genomes Project
The Mouse Genomes Project (https://www.sanger.ac.uk/science/data/mouse-genomes-project, last access: 3 January 2018) uses next-generation sequencing technologies to sequence the genomes of key laboratory mouse strains. The project consists of two branches: (1) short-read sequencing of many laboratory mouse strains and identification of sequence variation (SNPs, short insertions and deletions, and larger structural variations) relative to the C57BL/6J mouse reference genome. (2) De novo genome assembly and strain-specific gene annotation of the most widely used strains.
7.3 Mouse Gene Expression Database and Expression Atlas
The Mouse Gene Expression Database (Finger et al., 2017) contains information on gene expression in different inbred mice strains (http://www.informatics.jax.org/expression.shtml, last access: 3 January 2018). The gene expression atlas provides information on the temporal and spatial expression patterns of genes (https://www.ebi.ac.uk/gxa, last access: 3 January 2018). This allows researchers to find in which tissue and at which developmental stage a certain gene is expressed.
7.4 GeneNetwork
GeneNetwork (http://www.genenetwork.org, last access: 3 January 2018) is a free scientific web resource for mouse quantitative trait locus analysis. It is used to study the relationship between genes, environmental factors, phenotypes, and disease risk (Sloan et al., 2016; Wu et al., 2004). Starting out, it was the repository where genotype and phenotype information regarding BXD RIS was collected. Currently, in addition to BXD much more information is available for many different sets of RISs of mice including
-
AKXD derived from AKR/J and DBA/2J;
-
AXB/BXA, a reciprocal cross between A/J and C57BL/6J to investigate parent-of-origin effects;
-
BXH derived from a cross between a female C57BL/6J and a male C3H/HeJ;
-
CXB the oldest RIS cross (1971) derived from a cross between BALB/cBy and C57BL/6By;
-
LXS, a RIS with an intriguing history (it is derived from an eight-way cross initiated in the 1950s bred using a circle breeding method using the following mouse strains: A, AKR, BALB/c, C3H/2, C57BL, DBA/2, IS/Bi, and RIII).
In addition, phenotype and genotype data are also available on different cross types, such as F2 crosses (B6BTBRF2, B6D2F2, BH/HB F2, CastB6/B6Cast F2, B6JxB6N F2), but also on more complex outbred crosses such as the HS, the CC, and the Hybrid Mouse Diversity Panel. Recently, data from other species has also been integrated into GeneNetwork (human, rat, monkey, fruit flies, and others) to facilitate the translational research of results into other species. To this end, GeneNetwork provides many tools for the analysis of phenotype and genotype data as well as tools for association analyses in inbred and outbred populations.
Without any doubt, direct research in the target livestock species is unbeatable. In this regard, physiological, endocrine, and anatomical differences behind complex traits such as productivity, fertility, and behavior between species, in particular between the mouse and livestock species, must be considered. Such significant differences exist, for example, in the nutrient uptake and the metabolism between monogastric and ruminant animals and in the reproduction between unipara and multipara. Some genes, which are important for livestock, do not even exist in the mouse genome, for example genes involved in horn development. Nevertheless, although differences in the regulation of genes and signaling cascades exist, major gene functions and pathways are shared between all mammals. Therefore, mouse models can be used in comparative genetics and genomics to accelerate research in other species.
Livestock populations that are under selection for genetic improvement are often well characterized for genetic association studies. In such populations, pedigree information, reliable phenotypes for production, reproduction, health, and increasingly also for fitness traits, and dense genetic information are available. These are excellent prerequisites for successful mapping of genomic loci directly associated with causal genes in livestock. However, the subsequent gene identification and, moreover, finding the causal sequence variant, even if we expect different mutations in other species, remains a challenge due to linkage disequilibrium. Exceptions are defect and lethal genes, which can be disentangled through studies in affected livestock populations showing Mendelian co-segregation or missing homozygosity, respectively.
The special value of the mouse as a model animal for livestock research lies primarily in the existence of diverse genetic resources, the easy-to-handle genome, and comprehensive phenotypic measurements under a defined environment. Unique genetic resources such as inbred strains on the one hand and outbred strains with known genetic origin on the other hand allow the investigation of diverse complex traits, which are also of interest for livestock breeders. Structured multi-parental populations combine the higher statistical power of mapping quantitative trait loci with the high resolution of genome-wide association studies, allowing the almost direct identification of causal variants underlying the phenotype variation. Using these resources, the mouse became a powerful model animal for linkage and genome-wide association studies to discover or to finely map genetic loci affecting phenotype variation in complex traits. This allows translational information to flow from mouse studies into livestock research. In addition to direct genetic effects, imprinting and other epigenetic effects, the structure of complex traits, allelic imbalance, allele transmission disequilibria, and the inheritance of correlated traits have been discovered and quantified in mice.
Key tools, techniques, and protocols for the manipulation of the genome have been developed in mice. These include gene transfer, gene targeting, and genome editing as well as stem cell generation, differentiation, and manipulation. Using the different methods, the mouse largely contributes to functionally annotating genes and regulatory units. Genetically modified mice are excellent resources to study gene functions on different genetic backgrounds and under different challenging conditions. Through gene editing, modified mice can be quickly generated. Combined with the short generation interval this leads to the situation in which a new hypothesis about how a certain mutation will affect gene function can be formulated and tested within the span of a year. In many livestock species this would take much longer since the generation interval is orders of magnitude larger compared to mice. Moreover, the technical developments in mouse research contributed fundamentally to the progress in genome research in different species.
Finally, there might already be a mouse model available for a particular disease or phenotype of interest. This mouse model might give researches a hint on functional effects of their candidate gene or genes or confirm findings in their target species. Researchers can look up their candidate gene in diverse public databases or just order the mouse. Many gene-driven mouse models have been generated in the past 20 years and many of those are readily available from commercial distributers or research institutes.
Only publicly available data sets were used in this article.
The authors declare that they have no conflict of interest.
This study was supported by a grant from the German Research Foundation
(Deutsche Forschungsgemeinschaft, DFG).
Edited by: Steffen Maak
Reviewed by: two anonymous referees
Agarwal, R., Rout, P. K., and Singh, S. K.: Leptin: A biomolecule for enhancing livestock productivity, Indian J. Biotechnol., 8, 169–176, 2009.
Al-Asmakh, M. and Zadjali, F.: Use of germ-free animal models in microbiota-related research, J. Microbiol. Biotechnol., 25, 1583–1588, https://doi.org/10.4014/jmb.1501.01039, 2015.
Antoine-Moussiaux, N., Magez, S., and Desmecht, D.: Contributions of experimental mouse models to the understanding of African trypanosomiasis, Trends Parasitol., 24, 411–418, https://doi.org/10.1016/j.pt.2008.05.010, 2008.
Arends, D., Prins, P., Jansen, R. C., and Broman, K. W.: R/qtl: high-throughput multiple QTL mapping, Bioinformatics, 26, 2990–2992, https://doi.org/10.1093/bioinformatics/btq565, 2010.
Arends, D., Heise, S., Karst, S., Trost, J., and Brockmann, G. A.: Fine mapping a major obesity locus (jObes1) using a Berlin Fat Mouse × B6N advanced intercross population, Int. J. Obes., 40, 1784–1788, https://doi.org/10.1038/ijo.2016.150, 2016.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G.: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nat. Genet., 25, 25–29, https://doi.org/10.1038/75556, 2000.
Bailey, D. W.: Recombinant-inbred strains: An Aid to Finding Identity, Linkage, and Function of Histocompatibility and Other Genes, Transplantation, 11, 325–327, https://doi.org/10.1097/00007890-197103000-00013, 1971.
Baron, T. G. M., Biacabe, A. G., Bencsik, A., and Langeveld, J. P. M.: Transmission of new bovine prion to mice, Emerg. Infect. Dis., 12, 1125–1128, https://doi.org/10.3201/eid1207.060107, 2006.
Baron, T. G. M., Bencsik, A., Biacabe, A. G., Morignat, E., and Bessen, R. A.: Phenotypic similarity of transmissible mink encephalopathy in cattle and L-type bovine spongiform encephalopathy in a mouse model, Emerg. Infect. Dis., 13, 1887–1894, https://doi.org/10.3201/eid13112.070635, 2007.
Blom-Potar, M. C., Chamond, N., Cosson, A., Jouvion, G., Droin-Bergère, S., Huerre, M., and Minoprio, P.: Trypanosoma vivax infections: Pushing ahead with mouse models for the study of nagana. ii. immunobiological dysfunctions, PLoS Negl. Trop. Dis., 4, e793, https://doi.org/10.1371/journal.pntd.0000793, 2010.
Brockmann, G. A.: Inbreeding and corssbreeding, in: The Mouse in Animal Genetics and Breeding Research, Imperial College Press and distributed by World Scientific Publishing Co., Imperial College Press, Covent Garden, London, UK, 57–83, 2005.
Brockmann, G. A. and Bevova, M. R.: Using mouse models to dissect the genetics of obesity, Trends Genet., 18, 367–376, https://doi.org/10.1016/S0168-9525(02)02703-8, 2002.
Broman, K. W., Wu, H., Sen, S., and Churchill, G. A.: R/qtl: QTL mapping in experimental crosses, Bioinformatics, 19, 889–890, https://doi.org/10.1093/bioinformatics/btg112, 2003.
Brown, S. D. M. and Moore, M. W.: The International Mouse Phenotyping Consortium: Past and future perspectives on mouse phenotyping, Mamm. Genome, 23, 632–640, https://doi.org/10.1007/s00335-012-9427-x, 2012.
Bryant, C. D., Zhang, N. N., Sokoloff, G., Fanselow, M. S., Ennes, H. S., Palmer, A. A., and McRoberts, J. A.: Behavioral differences among C57BL/6 substrains: Implications for transgenic and knockout studies, J. Neurogenet., 22, 315–331, https://doi.org/10.1080/01677060802357388, 2008.
Charleston, B., Bankowski, B. M., Gubbins, S., Chase-Topping, M. E., Schley, D., Howey, R., Barnett, P. V, Gibson, D., Juleff, N. D., and Woolhouse, M. E. J.: Relationship between clinical signs and transmission of an infectious disease and the implications for control, Science, 332, 726–729, https://doi.org/10.1126/science.1199884, 2011.
Chen, X., Yang, J., Ji, Y., Okoth, E., Liu, B., Li, X., Yin, H., and Zhu, Q.: Recombinant Newcastle disease virus expressing African swine fever virus protein 72 is safe and immunogenic in mice, Virol. Sin., 31, 150–159, https://doi.org/10.1007/s12250-015-3692-2, 2016.
Chua, S. C., Chung, W. K., Wu-Peng, X. S., Zhang, Y., Liu, S. M., Tartaglia, L., and Leibel, R. L.: Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor, Science, 271, 994–996, https://doi.org/10.1126/science.271.5251.994, 1996.
Clavel, T., Lagkouvardos, I., Blaut, M., and Stecher, B.: The mouse gut microbiome revisited: From complex diversity to model ecosystems, Int. J. Med. Microbiol., 306, 316–327, https://doi.org/10.1016/j.ijmm.2016.03.002, 2016.
Clop, A., Marcq, F., Takeda, H., Pirottin, D., Tordoir, X., Bibé, B., Bouix, J., Caiment, F., Elsen, J. M., Eychenne, F., Larzul, C., Laville, E., Meish, F., Milenkovic, D., Tobin, J., Charlier, C., and Georges, M.: A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep, Nat. Genet., 38, 813–818, https://doi.org/10.1038/ng1810, 2006.
Complex Trait Consortium: The Collaborative Cross, a community resource for the genetic analysis of complex traits, Nat. Genet., 36, 1133–1137, 2004.
Cordes, S. P.: N-ethyl-N-nitrosourea mutagenesis: boarding the mouse mutant express, Microbiol. Mol. Biol. Rev., 69, 426–439, https://doi.org/10.1128/MMBR.69.3.426-439.2005, 2005.
Darvasi, A. and Soller, M.: Advanced intercross lines, an experimental population for fine genetic mapping, Genetics, 141, 1199–1207, 1995.
DeVuyst, E. A., Bullinger, J. R., Bauer, M. L., Berg, P. T., and Larson, D. M.: An economic analysis of genetic information: Leptin genotyping in fed cattle, J. Agric. Resour. Econ., 32, 291–305, 2007.
Dogan, A., Lasch, P., Neuschl, C., Millrose, M. K., Alberts, R., Schughart, K., Naumann, D., and Brockmann, G. A.: ATR-FTIR spectroscopy reveals genomic loci regulating the tissue response in high fat diet fed BXD recombinant inbred mouse strains, BMC Genomics, 14, 386, https://doi.org/10.1186/1471-2164-14-386, 2013.
Elsik, C. G., Tellam, R. L., and Worley, K. C.: The Genome Sequence of Taurine Cattle: A window to ruminant biology and evolution, Science, 324, 522–528, https://doi.org/10.1126/science.1169588, 2009.
Esvelt, K. M., Mali, P., Braff, J. L., Moosburner, M., Yaung, S. J., and Church, G. M.: Orthogonal Cas9 proteins for RNA-guided gene regulation and editing, Nat. Methods, 10, 1116–1123, https://doi.org/10.1038/nmeth.2681, 2013.
Evsikov, A. V., Dolan, M. E., Genrich, M. P., Patek, E., and Bult, C. J.: MouseCyc: A curated biochemical pathways database for the laboratory mouse, Genome Biol., 10, R84, https://doi.org/10.1186/gb-2009-10-8-r84, 2009.
Finger, J. H., Smith, C. M., Hayamizu, T. F., McCright, I. J., Xu, J., Law, M., Shaw, D. R., Baldarelli, R. M., Beal, J. S., Blodgett, O., Campbell, J. W., Corbani, L. E., Lewis, J. R., Forthofer, K. L., Frost, P. J., Giannatto, S. C., Hutchins, L. N., Miers, D. B., Motenko, H., Stone, K. R., Eppig, J. T., Kadin, J. A., Richardson, J. E., and Ringwald, M.: The mouse Gene Expression Database (GXD): 2017 update, Nucl. Acids Res., 45, D730–D736, https://doi.org/10.1093/nar/gkw1073, 2017.
Flowers, S. A. and Ellingrod, V. L.: The Microbiome in Mental Health: Potential Contribution of Gut Microbiota in Disease and Pharmacotherapy Management, Pharmacotherapy, 35, 910–916, https://doi.org/10.1002/phar.1640, 2015.
Foote, S. J., Iraqi, F., and Kemp, S. J.: Controlling malaria and African trypanosomiasis: The role of the mouse, Brief. Funct. Genom. Proteom., 4, 214–224, https://doi.org/10.1093/bfgp/4.3.214, 2005.
Fürbass, R., Winter, A., Fries, R., and Kühn, C.: Alleles of the bovine DGAT1 variable number of tandem repeat associated with a milk fat QTL at chromosome 14 can stimulate gene expression, Physiol. Genomics, 25, 116–120, https://doi.org/10.1152/physiolgenomics.00145.2005, 2006.
Gatti, D. M., Svenson, K. L., Shabalin, A., Wu, L.-Y., Valdar, W., Simecek, P., Goodwin, N., Cheng, R., Pomp, D., Palmer, A., Chesler, E. J., Broman, K. W., and Churchill, G. A.: Quantitative Trait Locus Mapping Methods for Diversity Outbred Mice, G3 Genes Genom. Genet., 4, 1623–1633, https://doi.org/10.1534/g3.114.013748, 2014.
Gene Ontology Consortium: Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium, Nucl. Acids Res., 45, D331–D338, https://doi.org/10.1093/nar/gkw1108, 2017.
Giblin, L., Butler, S. T., Kearney, B. M., Waters, S. M., Callanan, M. J., and Berry, D. P.: Association of bovine leptin polymorphisms with energy output and energy storage traits in progeny tested Holstein-Friesian dairy cattle sires, BMC Genet., 11, 73, https://doi.org/10.1186/1471-2156-11-73, 2010.
Goddard, M. E., Kemper, K. E., MacLeod, I. M., Chamberlain, A. J., and Hayes, B. J.: Genetics of complex traits: prediction of phenotype, identification of causal polymorphisms and genetic architecture, P. Roy. Soc. B, 283, 20160569, https://doi.org/10.1098/rspb.2016.0569, 2016.
Grisart, B., Coppieters, W., Farnir, F., Karim, L., Ford, C., Berzi, P., Cambisano, N., Mni, M., Reid, S., Simon, P., Spelman, R., Georges, M., and Snell, R.: Positional candidate cloning of a QTL in dairy cattle: Identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition, Genome Res., 12, 222–231, https://doi.org/10.1101/gr.224202, 2002.
Grobet, L., Martin, L. J. R., Poncelet, D., Pirottin, D., Brouwers, B., Riquet, J., Schoeberlein, A., Dunner, S., Ménissier, F., Massabanda, J., Fries, R., Hanset, R., and Georges, M.: A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle, Nat. Genet., 17, 71–74, https://doi.org/10.1038/ng0997-71, 1997.
Guo, R., Wan, Y., Xu, D., Cui, L., Deng, M., Zhang, G., Jia, R., Zhou, W., Wang, Z., Deng, K., Huang, M., Wang, F., and Zhang, Y.: Generation and evaluation of Myostatin knock-out rabbits and goats using CRISPR/Cas9 system, Sci. Rep., 6, 29855, https://doi.org/10.1038/srep29855, 2016.
Gupta, R. M. and Musunuru, K.: Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9, J. Clin. Invest., 124, 4154–4161, https://doi.org/10.1172/JCI72992, 2014.
Habiela, M., Seago, J., Perez-Martin, E., Waters, R., Windsor, M., Salguero, F. J., Wood, J., Charleston, B., and Juleff, N.: Laboratory animal models to study foot-and-mouth disease: a review with emphasis on natural and vaccine-induced immunity, J. Gen. Virol., 95, 2329–2345, https://doi.org/10.1099/vir.0.068270-0, 2014.
Hitzemann, R., Malmanger, B., Cooper, S., Coulombe, S., Reed, C., Demarest, K., Koyner, J., Cipp, L., Flint, J., Talbot, C., Rademacher, B., Buck, K., and McCaughran, J.: Multiple cross mapping (MCM) markedly improves the localization of a QTL for ethanol-induced activation, Genes. Brain. Behav., 1, 214–222, https://doi.org/10.1034/j.1601-183X.2002.10403.x, 2002.
Humphray, S. J., Scott, C. E., Clark, R., Marron, B., Bender, C., Camm, N., Davis, J., Jenks, A., Noon, A., Patel, M., Sehra, H., Yang, F., Rogatcheva, M. B., Milan, D., Chardon, P., Rohrer, G., Nonneman, D., de Jong, P., Meyers, S. N., Archibald, A., Beever, J. E., Schook, L. B., and Rogers, J.: A high utility integrated map of the pig genome, Genome Biol., 8, R139, https://doi.org/10.1186/gb-2007-8-7-r139, 2007.
Iannuzzi, L., Di Meo, G. P., Perucatti, A., and Incarnato, D.: Comparison of the human with the sheep genomes by use of human chromosome-specific painting probes, Mamm. Genome, 10, 719–723, https://doi.org/10.1007/s003359901078, 1999.
Kambadur, R., Sharma, M., Smith, T. P. L., and Bass, J. J.: Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle, Genome Res., 7, 910–916, https://doi.org/10.1101/gr.7.9.910, 1997.
Kinross, J. M., Darzi, A. W., and Nicholson, J. K.: Gut microbiome-host interactions in health and disease, Genome Med., 3, 14, https://doi.org/10.1186/gm228, 2011.
Kononoff, P. J., Deobald, H. M., Stewart, E. L., Laycock, A. D., and Marquess, F. L. S.: The effect of a leptin single nucleotide polymorphism on quality grade, yield grade, and carcass weight of beef cattle, J. Anim. Sci., 83, 927–932, https://doi.org/10.2527/2005.834927x, 2005.
Lyon, M. F.: Rules and guidelines for gene nomenclature, in: 2nd Edn., edited by: Searle, A. G., Oxford University Press, Oxford, 1989.
McPherron, A. C. and Lee, S.-J.: Double muscling in cattle due to mutations in the myostatin gene, P. Natl. Acad. Sci. USA, 94, 12457–12461, https://doi.org/10.1073/pnas.94.23.12457, 1997.
McPherron, A. C., Lawler, A. M., and Lee, S. J.: Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member, Nature, 387, 83–90, https://doi.org/10.1038/387083a0, 1997.
Mekada, K., Abe, K., Murakami, A., Nakamura, S., Nakata, H., Moriwaki, K., Obata, Y., Yoshiki, A., and Yoshiki, A.: Genetic Differences among C57BL/6 Substrains, Exp. Anim., 58, 141–149, https://doi.org/10.1538/expanim.58.141, 2009.
Nguyen, N. Y. N., Maxwell, M. J., Ooms, L. M., Davies, E. M., Hilton, A. A., Collinge, J. E., Hilton, D. J., Kile, B. T., Mitchell, C. A., Hibbs, M. L., Jane, S. M., and Curtis, D. J.: An ENU-induced mouse mutant of SHIP1 reveals a critical role of the stem cell isoform for suppression of macrophage activation, Blood, 117, 5362–5371, https://doi.org/10.1182/blood-2011-01-331041, 2011.
Nolan, P. M., Hugill, A., and Cox, R. D.: ENU mutagenesis in the mouse: Application to human genetic disease, Brief. Funct. Genom. Proteom., 1, 278–289, https://doi.org/10.1093/bfgp/1.3.278, 2002.
Oreper, D., Cai, Y., Tarantino, L. M., de Villena, F. P.-M., and Valdar, W.: Inbred Strain Variant Database (ISVdb): A Repository for Probabilistically Informed Sequence Differences Among the Collaborative Cross Strains and Their Founders, G3 Genes Genom. Genet., 7, 1623–1630, https://doi.org/10.1534/g3.117.041491, 2017.
Rabot, S., Membrez, M., Bruneau, A., Gerard, P., Harach, T., Moser, M., Raymond, F., Mansourian, R., and Chou, C. J.: Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism, FASEB J., 24, 4948–4959, https://doi.org/10.1096/fj.10-164921, 2010.
Revilla, Y., Pena, L., Mampaso, F., Viñuela, E., and Martinez-Alonso, C.: Swine-reconstituted SCID mice as a model for African swine fever virus infection, J. Gen. Virol., 75, 1983–1988, https://doi.org/10.1099/0022-1317-75-8-1983, 1994.
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wüthrich, K.: NMR structure of the mouse prion protein domain PrP(121–231), Nature, 382, 180–182, https://doi.org/10.1038/382180a0, 1996.
Rjeibi, M. R., Ben Hamida, T., Dalgatova, Z., Mahjoub, T., Rejeb, A., Dridi, W., and Gharbi, M.: First report of surra (Trypanosoma evansi infection) in a Tunisian dog, Parasite, 22, 3, https://doi.org/10.1051/parasite/2015004, 2015.
Rosshart, S. P., Vassallo, B. G., Angeletti, D., Hutchinson, D. S., Morgan, A. P., Takeda, K., Hickman, H. D., McCulloch, J. A., Badger, J. H., Ajami, N. J., Trinchieri, G., Pardo-Manuel de Villena, F., Yewdell, J. W., and Rehermann, B.: Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance, Cell, 171, 1015–1028, https://doi.org/10.1016/j.cell.2017.09.016, 2017.
Schering, L., Hoene, M., Kanzleiter, T., Jähnert, M., Wimmers, K., Klaus, S., Eckel, J., Weigert, C., Schürmann, A., Maak, S., Jonas, W., and Sell, H.: Identification of novel putative adipomyokines by a cross-species annotation of secretomes and expression profiles, Arch. Physiol. Biochem., 24, 1–12, https://doi.org/10.3109/13813455.2015.1092044, 2015.
Schibler, L., Vaiman, D., Oustry, A., Giraud-Delville, C., and Cribiu, E. P.: Comparative gene mapping: A fine-scale survey of chromosome rearrangements between ruminants and humans, Genome Res., 8, 901–915, https://doi.org/10.1101/gr.8.9.901, 1998.
Schmid, M. and Bennewitz, J.: Invited review: Genome-wide association analysis for quantitative traits in livestock – a selective review of statistical models and experimental designs, Arch. Anim. Breed., 60, 335–346, https://doi.org/10.5194/aab-60-335-2017, 2017.
Scott, M. R., Safar, J., Telling, G., Nguyen, O., Groth, D., Torchia, M., Koehler, R., Tremblay, P., Walther, D., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B.: Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice, P. Natl. Acad. Sci. USA, 94, 14279–14284, https://doi.org/10.1073/pnas.94.26.14279, 1997.
Sherry, S. T.: dbSNP: the NCBI database of genetic variation, Nucl. Acids Res., 29, 308–311, https://doi.org/10.1093/nar/29.1.308, 2001.
Síma, M., Havelková, H., Quan, L., Svobodová, M., Jarošíková, T., Vojtíšková, J., Stassen, A. P. M., Demant, P., and Lipoldová, M.: Genetic control of resistance to Trypanosoma brucei brucei infection in mice, PLoS Negl. Trop. Dis., 5, e1173, https://doi.org/10.1371/journal.pntd.0001173, 2011.
Skinner, H. H.: Propagation of Strains of Foot-and-Mouth Disease Virus in Unweaned White Mice, P. Roy. Soc. Med., 44, 1041–1044, 1951.
Sloan, Z., Arends, D., Broman, K. W., Centeno, A., Furlotte, N., Nijveen, H., Yan, L., Zhou, X., Williams, R. W., and Prins, P.: GeneNetwork: framework for web-based genetics, J. Open Source Softw., 1, 8–10, https://doi.org/10.21105/joss.00025, 2016.
Smith, S. J., Cases, S., Jensen, D. R., Chen, H. C., Sande, E., Tow, B., Sanan, D. A., Raber, J., Eckel, R. H., and Farese, R. V.: Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat, Nat. Genet., 25, 87–90, https://doi.org/10.1038/75651, 2000.
Svenson, K. L., Von Smith, R., Magnani, P. A., Suetin, H. R., Paigen, B., Naggert, J. K., Li, R., Churchill, G. A., and Peters, L. L.: Multiple trait measurements in 43 inbred mouse strains capture the phenotypic diversity characteristic of human populations, J. Appl. Physiol., 102, 2369–2378, https://doi.org/10.1152/japplphysiol.01077.2006, 2007.
Telling, G. C.: Transgenic mouse models and prion strains, Top. Curr. Chem., 305, 79–100, https://doi.org/10.1007/128_2011_166, 2011.
Thaller, G., Krämer, W., Winter, A., Kaupe, B., Erhardt, G., and Fries, R.: Effects of DGAT1 variants on milk production traits in German cattle breeds, J. Anim. Sci., 81, 1911–1918, 2003.
Tulman, E. R., Delhon, G. A., Ku, B. K., and Rock, D. L.: African swine fever virus, Curr. Top. Microbiol. Immunol., 328, 43–87, https://doi.org/10.1007/978-3-540-68618-7_2, 2009.
Wang, K., Tang, X., Xie, Z., Zou, X., Li, M., Yuan, H., Guo, N., Ouyang, H., Jiao, H., and Pang, D.: CRISPR/Cas9-mediated knockout of myostatin in Chinese indigenous Erhualian pigs, Transgenic Res., 26, 799–805, https://doi.org/10.1007/s11248-017-0044-z, 2017.
Williams, J. L.: Genetic control of meat quality traits, in: Meat Biotechnology, Springer, New York, NY, 21–60, 2008.
Wilson, D. E. and Reeder, D. M.: Mammal Species of the World. A Taxonomic and Geographic Reference, in: 3rd Edn., Johns Hopkins University Press, Baltimore, Maryland, USA, 2005.
Wilson, R., Hart, P., Piccardo, P., Hunter, N., Casalone, C., Baron, T., and Barron, R. M.: Bovine PrP expression levels in transgenic mice influence transmission characteristics of atypical bovine spongiform encephalopathy, J. Gen. Virol., 93, 1132–1140, https://doi.org/10.1099/vir.0.040030-0, 2012.
Wu, C., Huang, H., Juan, H., and Chen, S.: GeneNetwork: an interactive tool for reconstruction of genetic networks using microarray data, Bioinformatics, 20, 3691–3693, https://doi.org/10.1093/bioinformatics/bth428, 2004.
Yalcin, B., Willis-Owen, S. A. G., Fullerton, J., Meesaq, A., Deacon, R. M., Rawlins, J. N. P., Copley, R. R., Morris, A. P., Flint, J., and Mott, R.: Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice, Nat. Genet., 36, 1197–1202, https://doi.org/10.1038/ng1450, 2004.
Zerbino, D. R., Achuthan, P., Akanni, W., Amode, M. R., Barrell, D., Bhai, J., Billis, K., Cummins, C., Gall, A., Girón, C. G., Gil, L., Gordon, L., Haggerty, L., Haskell, E., Hourlier, T., Izuogu, O. G., Janacek, S. H., Juettemann, T., To, J. K., Laird, M. R., Lavidas, I., Liu, Z., Loveland, J. E., Maurel, T., McLaren, W., Moore, B., Mudge, J., Murphy, D. N., Newman, V., Nuhn, M., Ogeh, D., Ong, C. K., Parker, A., Patricio, M., Riat, H. S., Schuilenburg, H., Sheppard, D., Sparrow, H., Taylor, K., Thormann, A., Vullo, A., Walts, B., Zadissa, A., Frankish, A., Hunt, S. E., Kostadima, M., Langridge, N., Martin, F. J., Muffato, M., Perry, E., Ruffier, M., Staines, D. M., Trevanion, S. J., Aken, B. L., Cunningham, F., Yates, A., and Flicek, P.: Ensembl 2018, Nucl. Acids Res., 46, D754–D761, https://doi.org/10.1093/nar/gkx1098, 2017.
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M.: Positional cloning of the mouse obese gene and its human homologue, Nature, 372, 425–432, https://doi.org/10.1038/372425a0, 1994.
- Abstract
- Introduction
- The necessity of using knowledge from model organisms in livestock research
- The mouse as a model animal for livestock research
- Diversity among laboratory mice
- Functional gene annotation through genetic modification
- Exemplary mouse model contributions to livestock research
- Mouse specific databases
- Concluding remarks
- Data availability
- Competing interests
- Acknowledgements
- References
- Abstract
- Introduction
- The necessity of using knowledge from model organisms in livestock research
- The mouse as a model animal for livestock research
- Diversity among laboratory mice
- Functional gene annotation through genetic modification
- Exemplary mouse model contributions to livestock research
- Mouse specific databases
- Concluding remarks
- Data availability
- Competing interests
- Acknowledgements
- References